DeepMind开源AlphaFold,蛋白质预测模型登上《Nature》


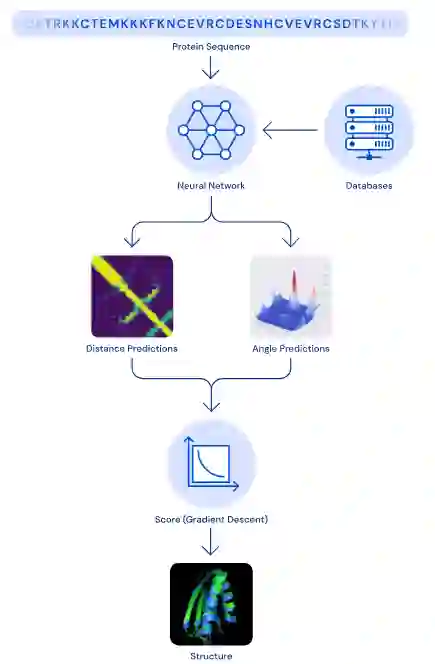
模型简介

https://www.biorxiv.org/content/10.1101/846279v1.full.pdf
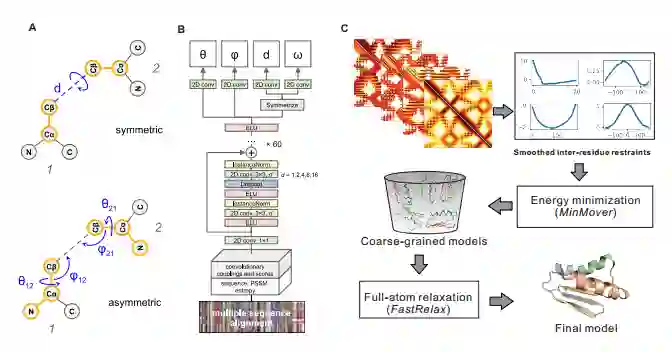
图注:A:用角度和距离表示从一个残基到另一残基的转换。B:神经网络结构根据MSA预测残基间的几何形状;C:预测过程概览
 ~之间的距离之外,残基对之间的方位也属于其预测的范围。如上图所示,残基1与残基2之间的方位由3个二面角
~之间的距离之外,残基对之间的方位也属于其预测的范围。如上图所示,残基1与残基2之间的方位由3个二面角 以及2个平面角
以及2个平面角 表示。其中ω表示沿虚轴(连接两个残基的原子)旋转角度。
表示。其中ω表示沿虚轴(连接两个残基的原子)旋转角度。  角度定义了从残基1看到残基2的Cβ原子的方向,
角度定义了从残基1看到残基2的Cβ原子的方向, 同理。另外,与d和ω不同,θ和φ坐标是不对称的,其取决于残基的顺序。综上6个参数d,ω,
同理。另外,与d和ω不同,θ和φ坐标是不对称的,其取决于残基的顺序。综上6个参数d,ω, 定义了两个残基的主干原子的相对位置,这6个参数也是神经网络所要预测的。
定义了两个残基的主干原子的相对位置,这6个参数也是神经网络所要预测的。
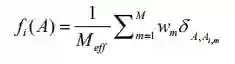
 是MSA中序列数目的倒数,与序列m至少有80%的序列同源性。其中
是MSA中序列数目的倒数,与序列m至少有80%的序列同源性。其中
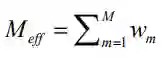
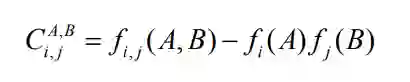
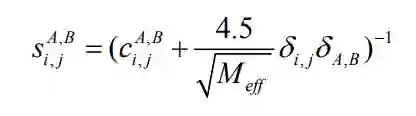
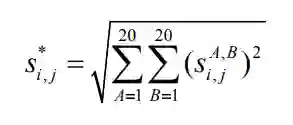
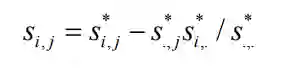
 分别为矩阵
分别为矩阵 的行列以及平均值。
的行列以及平均值。
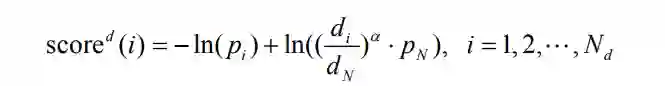
 是第i个bin的距离,
是第i个bin的距离, 是第i个bin的距离的概率,N是bin的总数。
是第i个bin的距离的概率,N是bin的总数。
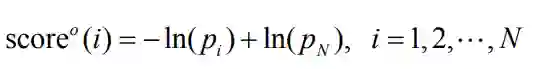
https://github.com/deepmind/deepmind-research/tree/master/alphafold_casp13



登录查看更多
相关内容
专知会员服务
20+阅读 · 2020年5月14日
Arxiv
59+阅读 · 2020年1月20日
Arxiv
4+阅读 · 2018年11月21日

相关VIP内容
专知会员服务
20+阅读 · 2020年5月14日
相关资讯
相关论文
Arxiv
59+阅读 · 2020年1月20日
Arxiv
4+阅读 · 2018年11月21日