
编译|王娜 审稿|王海云
本文介绍由美国德克萨斯大学MD安德森癌症中心生物统计学系的Ziyi Li和Kim-Anh Do共同通讯发表在 Bioinformatics 的研究成果:为了更好地注释scRNA-seq 数据,发现新的细胞类型,作者开发了一种简单而有效的方法,结合自动编码器和迭代特征选择,从scRNA-seq数据中自动识别新细胞。该方法用标记的训练数据训练一个自动编码器,并将自动编码器应用于测试数据以获得重建误差。通过反复选择表现出双模模式的特征,并使用所选特征对细胞进行重新分组,该方法可以准确地识别训练数据中不存在的新细胞。作者进一步将这种方法与支持向量机结合起来,为注释所有的细胞类型提供了一个完整的解决方案。使用五个真实的scRNA-seq数据集进行的广泛的数值实验,结果表明,该方法比现有的方法具有更好的性能。
简介
单细胞RNA测序(scRNA-seq)的出现使研究人员能够以前所未有的精度和准确度研究人类组织的细胞组成和转录组概况。作为第一步,注释细胞和分配细胞类型标签是最重要的步骤之一,因为大多数的下游分析都依赖于细胞标签的准确性。由于不同的研究人员对细胞类型标志物的理解存在差异,通常不能保证注释结果的可重复性。尽管多种监督方法为细胞注释提供了各种解决方案,但监督方法面临的一个大挑战是如何将新的(或未知的)细胞类型与已知的细胞类型区分开来。
在这项工作中,作者开发了一种新的两步法来自动标记含有新细胞的scRNA-seq数据。称之为使用基于机器学习的方法对未知细胞的存在进行细胞注释(CAMLU)。在第一步,CAMLU使用自动编码器和迭代特征选择的组合来区分已知细胞类型和新的细胞类型。这样的目的是,用训练数据训练自动编码器后,自动编码器将包含所有已知细胞类型的信息。将这个自动编码器应用于测试数据将产生所有基因的重构误差。由于细胞是已知和未知细胞类型的混合体,一些 "有信息的 "基因在其重构误差中会有双模分布,代表它们与已知细胞类型的不同相似程度。通过迭代特征选择,CAMLU可以选择一组较小的信息性特征,这些特征在已知和未知细胞群中具有表达差异,并最终将新型细胞与已知细胞类型区分开来。然后,可以根据这些信息性基因重新对细胞进行分组,并识别出新型细胞。除去第一步中确定的新细胞,CAMLU在第二步中使用支持向量机对其余的细胞进行详尽的注释。图1展示了CAMLU的工作流程。
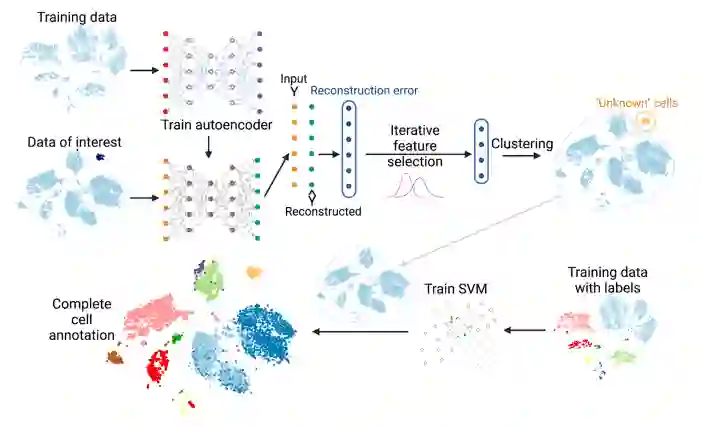
图1 CAMLU的工作流程
结果
蒙特卡洛数值实验 为了广泛地评估CAMLU的性能,作者设计了三个基于真实数据集的蒙特卡洛数值实验。作者将CAMLU与四种流行的细胞注释方法进行了比较,这些方法能够使用 "未分配 "的标签来识别未知的细胞。
用PBMC和HNCC细胞系进行的数值实验 PBMC数据有6万多个来自8种免疫细胞类型的分类细胞。HNCC总共有4632个癌细胞。对于每个实验,我们从PBMC数据中每个细胞类型随机选择个细胞,从HNCC数据中随机选择个癌症细胞。考虑了三种设置,正常细胞样本量=300;400;500(即训练数据中2400、3100和3800个细胞),对应于图2中的小、中和大。在所有设置中,癌细胞数量保持不变,即=300。图2总结了100多次蒙特卡洛实验的数值实验情况。与现有的方法相比,CAMLU在区分癌细胞和新型细胞以及标记全部细胞类型方面具有最高的准确性。CopyKAT在识别图2上图中的癌细胞方面是第二好的。由于它不能分配完整的标签列表,copyKAT没有在图2中展示。在其他现有的方法中,scmap-cluster和CHETAH也能很好地分配正确的标签,但准确率和ARI略低,其次是scPred。Scmap-cell在这两项任务中的准确率最低,可能是由于该方法产生了大量的 "未分配 "标签。
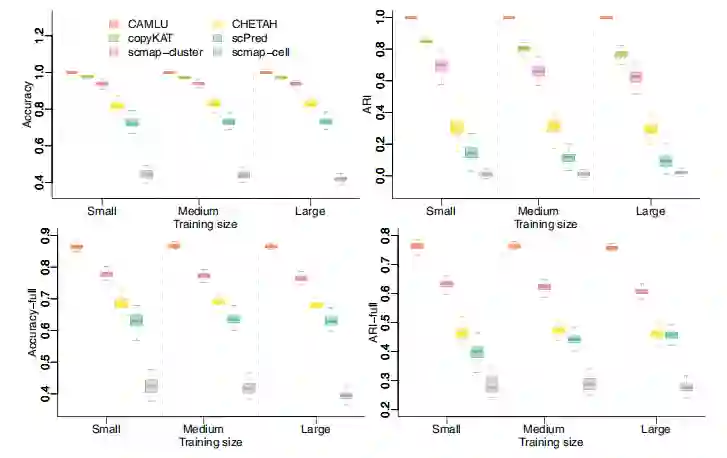
图2 使用PBMC数据和癌细胞系数据的混合物进行数值实验的结果
用PBMC进行的数值实验 接下来,作者设计了一个数值实验,只用PBMC数据来模拟未知细胞不是非整倍体时的情况。我们将单核细胞作为 "新型 "细胞类型,其他七种细胞类型作为已知细胞类型。与第一个实验类似,作者为其余七种细胞类型的每个细胞类型随机选择个细胞,从单核细胞中选择个细胞。作者再次考虑了三种设置,已知细胞类型的大小不同,=300;400;500,单核细胞的=300。
图3A中总结了CAMLU和现有方法的准确度。CAMLU在识别测试数据中的单核细胞和分配所有标签方面具有最高的准确性。作者发现scmap-cluster在这两项任务中都是第二好的方法,其次是CHETAH和scPred,性能相似。与第一次数值实验相比,CAMLU的准确率下降了一点,可能是因为目前的设置比较难。随着训练样本量的增加,所有方法的性能都略有提高。
图3B和C展示了单个实验中真实和估计标签的新型细胞识别结果,这可能阐明了CAMLU和现有方法的差异。CAMLU在区分单核细胞和已知细胞方面具有几乎完美的准确性,而现有的方法,特别是scmap-cell,往往将很多已知细胞标记为 "未分配"。
用胰腺数据进行的数值实验 除PBMC数据外,作者还获得了胰腺scRNAseq数据集,以进一步评估新细胞为二倍体时的情况。作者通过将间质细胞作为未知细胞类型来设计实验。对于每次模拟,从 "已知 "细胞类型中随机抽取500个非间质细胞,并将这些细胞的数据作为训练数据。其余的 "已知 "细胞与间充质细胞结合起来作为测试数据。其中间质细胞数量为80,训练数据中的细胞总数为1626。
在图4中总结了100个蒙特卡洛实验的结果。在图4A中,CAMLU与其他现有的方法相比,具有更高的识别精度,精度平均提高5-10%。在图4B中,作者将CAMLU和其他方法的新型细胞类型和整体注释的细胞标签与一次实验的真实标签进行了可视化对比。CAMLU在这两项任务中脱颖而出,在区分极少数的新型细胞(<5%的测试数据)时表现出很高的准确性。CHETAH在这种情况下也有良好的表现,scmap-cell排名第三。scmap-cluster和scPred都不能识别新型细胞。
图3 使用单核细胞作为新的细胞类型的PBMC数据进行数值实验的结果
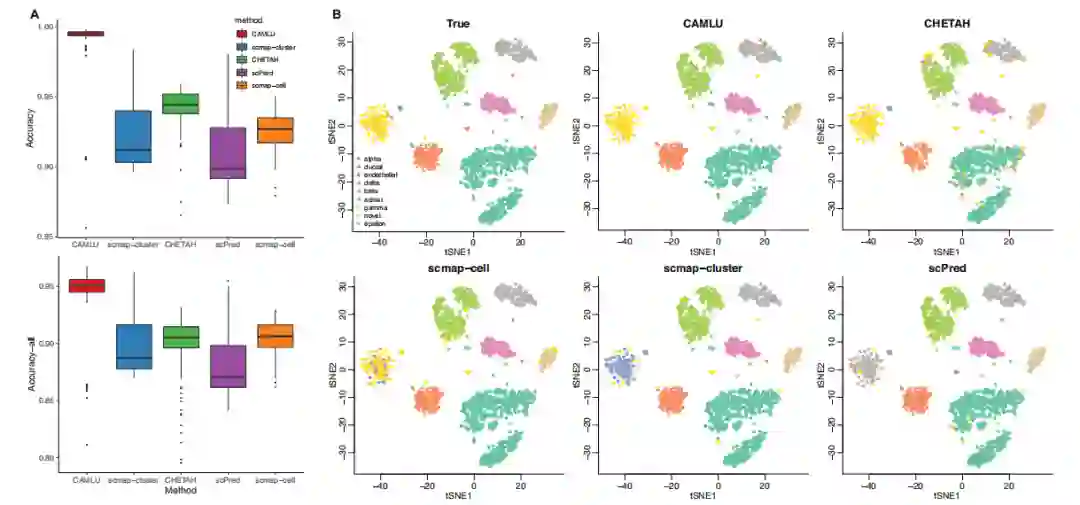
图4 使用胰腺数据的数值实验结果,以间质细胞为新型细胞类型
应用于两个真实的癌症数据集 在这个实验中,作者获得了一个scRNA-seq数据集,包括5名三阴性乳腺癌(TNBC)患者和另一个包括5名无性系甲状腺癌(ATC)患者的数据集。作者首先评估了所有方法从测试数据中识别恶性细胞的准确性。在图5B中,跨主题的细胞注释给分析带来了额外的噪声,与之前的设置相比,所有的方法都有较低的准确性。在所有的方法中,CAMLU仍然是该任务中最准确的方法,其平均准确率约为0.9。性能第二好的方法是在TNBC数据集的scmap-cluster和在ATC数据集的scPred。在TNBC数据中,scmap-cluster的准确率约为0.80,所有其他方法的平均准确率都低于0.6。对于ATC,所有现有的方法的准确率都在0.5-0.6左右。
在图5B中,作者说明了四个top基因在正常(蓝条)和恶性细胞(红条)中的重建误差分布。例如,COL6A2编码VI型胶原蛋白的三条α链之一,并被报道通过影响肿瘤和基质细胞来促进肿瘤的进展。作者发现COL6A2在恶性细胞中的重建误差比在正常细胞中高得多,表明COL6A2可能是两组细胞之间的差异基因。
图5C显示了使用TNBC和ATC数据从CAMLU中选出的前200个特征的最重要的Hallmark术语。作者在结果中发现了一些与疾病相关的术语。例如,在TNBC中,通过途径的信号传递是最重要的Hallmark术语。大量的现有研究报告了炎症因子TNF-a对乳腺癌生长的促进作用。同样,ATC结果中的首要术语,上皮-间质转化是与上皮性肿瘤进展、局部侵袭和转移有关的重要机制。一些研究报告指出,上皮-间质转化与ATC的进展密切相关。
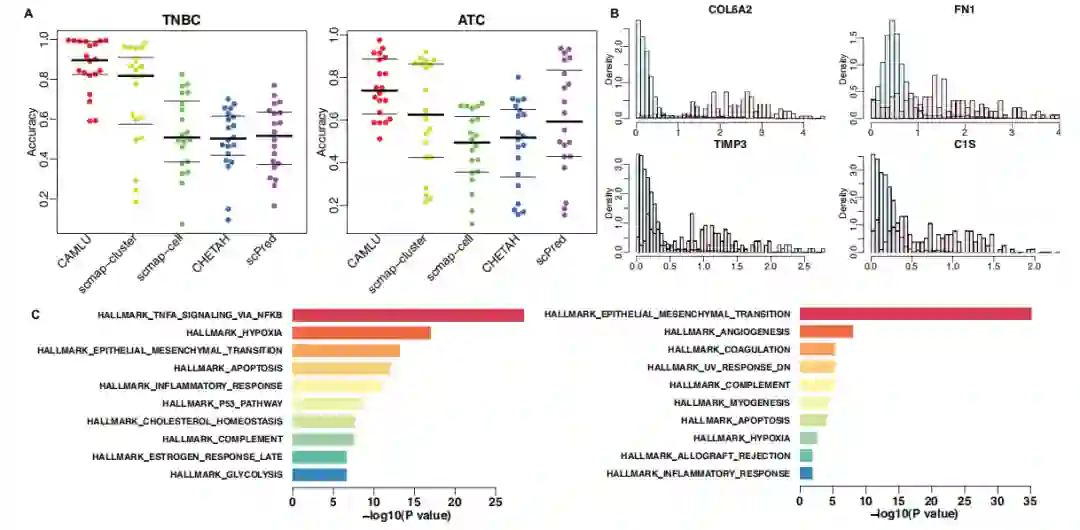
图5 在两个癌症数据集上应用CAMLU和现有方法的结果,即三阴性乳腺癌(TNBC)和非弹性甲状腺癌(ATC)
总结与讨论
在这项工作中,作者开发了一种基于机器学习的新方法,用于从scRNA-seq数据中识别未知细胞。该方法利用自动编码器和迭代特征选择的组合,根据信息特征的重建误差来识别新型细胞。在识别 "未知 "细胞后,其余的细胞使用支持向量机进行注释。与大多数将低相关度或低置信度的细胞标记为新型细胞的现有方法相比,该方法创新性地将未知细胞的选择和现有细胞类型的注释分开。
该有几个优点。首先,不依赖于非整倍体/二倍体细胞状态。第二,可以应用于识别不同大小的新型细胞。第三,尽管跨主体预测给问题带来了额外的噪音,并降低了所有方法的准确性,但该方法仍然比服务于类似目的的现有方法取得更高的性能。
有几个方向可以考虑和探索未来的工作。首先,作者将继续探索不同参数的选择,以提高方法的敏感性和稳健性。例如,细胞类型结构的不同复杂性可能需要更大或更小的自动编码器模型。选择的特征数量也可以与感兴趣的问题有关。可以设计自适应程序,在模型构建中自动选择这些因素。第二,可以考虑通过在框架中加入额外的生物知识,使工具更好地适应不同的疾病环境。在目前的特征选择设置中,作者仅仅根据重建分布来选择顶级特征。将双模特征与疾病相关的特征相结合,有可能获得更好的性能。
参考资料
Ziyi Li, Yizhuo Wang, Irene Ganan-Gomez, Simona Colla, Kim-Anh Do. A machine learning-based method for automatically identifying novel cells in annotating single-cell RNA-seq data. Bioinformatics; doi: https://doi.org/10.1093/bioinformatics/btac617
代码

