编译 | 程昭龙
审稿 | 林荣鑫,王静 本文介绍由同济大学控制科学与工程系的洪奕光和中国科学院数学与系统科学研究院的万林共同通讯发表在 Nature Communications 的研究成果:单细胞数据集成可以提供细胞的全面分子视图。然而,如何整合异质性单细胞多组学以及空间分辨的转录组学数据仍然是一个重大挑战。为此,作者提出了uniPort,这是一种结合耦合变分自动编码器(coupled-VAE)和小批量不平衡最优传输(Minibatch-UOT)的统一单细胞数据集成框架。它利用高度可变的通用基因和数据集特异性基因进行集成,以处理数据集之间的异质性,并可扩展到大规模数据集。uniPort 将异质性单细胞多组学数据集嵌入到共享的潜在空间。它还可以进一步构建一个用于跨数据集基因插补的参考图谱。同时,uniPort提供了一个灵活的标签传输框架,以使用最优传输计划去卷积异构的空间转录组数据,而不是嵌入潜在空间。作者通过应用uniPort集成多种数据集,包括单细胞转录组学、染色质可及性和空间分辨转录组学数据,从而证明了uniPort的能力。

简介
单细胞RNA测序(scRNA)和单细胞染色质转座酶可及性测序(scATAC)等高通量单细胞多组学测序技术的发展,能够对构成组织的异质性细胞群、发育过程的动力学以及控制细胞功能的潜在调节机制进行全面研究。单细胞数据集的计算集成是机器学习和数据科学领域的重要研究方向。
现有的单细胞集成方法中,大量工作致力于同时集成来自同一细胞的多个数据集。然而,这些配对数据集在技术上具有挑战性,且获取成本高昂。因此,针对来自相同或相似群体的不同细胞的数据,开发了大量的集成方法。例如,Seurat平台使用典型相关分析(CCA)将特征空间投影到公共子空间中,从而最大化数据集间的相关性。LIGER和DC3采用非负矩阵分解寻找共同特征的共享低维因子来匹配单细胞组学数据集。Harmony在最大多样性聚类和基于混合模型的线性批次校正之间进行协调迭代,提供了一个潜在空间来去除批次效应。然而,这些方法依赖于线性操作,因此缺乏处理跨细胞模态的非线性变形的能力。此外,它们只利用过滤后的常见基因,而忽略了数据集特异性基因对于识别细胞群的重要性,而这些细胞群通常能捕获不存在于常见基因中的细胞类型异质性。为了解决这些缺点,多种对比方法在集成单细胞多组学数据集方面取得了很有前景的结果。然而,流形对齐方法受到相对较高的计算复杂度限制,并且不能扩展到大规模数据集。
随着深度学习的发展,现已提出了许多基于自动编码器的方法,并证明了它们在跨模态数据集成中的能力。然而,其中大多数方法需要来自相同细胞的配对数据集以利用细胞配对信息,如DCCA和Cobolt。当细胞配对信息不可用时,替代方法是同时训练不同的自动编码器,并在潜在空间中跨不同的模态对齐细胞。最近,出现了大量的方法来解释非配对数据。例如,scDART和跨模态自动编码器通过自动编码器学习潜在空间,并通过基于核或基于判别器的差异来对齐潜在表示。然而,这些方法需要进行全局比对,这对于整合异质细胞群往往具有限制性。此外,已开发的基于迁移学习的方法,通过学习模态不变的潜在空间,将知识(如细胞标签)从一个模态迁移到另一个模态,从而建立源图谱。这虽然取得了较好的结果,但仅限于使用带有标注细胞标签的源模态。
最近发表的单细胞基因组学集成方法,如scMC和SCALEX,在一种模式的批次效应校正方面表现出了最佳性能,但它们尚未成为单细胞多组学数据集成的基准。GLUE是另一种最先进的单细胞多组学集成和集成调控推理方法,其开发了基于高级图的自动编码器。同时,许多其他方法被提出用于空间转录组学(ST)和scRNA数据的集成分析。其中,gimVI和Tangram取得了最优性能。然而,目前尚未开发出统一集成单细胞多组学以及空间分辨转录组学数据的方法。
为此,作者提出了uniPort,这是一个精确、稳健和高效的计算平台,用于将异构单细胞数据集与最佳传输(OT)集成。为了克服传统VAE在单细胞异质性或非配对数据集成方面的局限性,作者提出了一个通过结合耦合变分自动编码器(coupled-VAE)和小批量不平衡最优传输 (Minibatch-UOT) 的统一计算框架(图1)。该框架允许利用高度可变的常见基因和数据集特异性基因进行集成,以处理不同数据集的异质性。实验结果表明,uniPort可以准确、稳健地集成来自外周血单核细胞(PBMC)和小鼠脾脏的scATAC和scRNA数据集。它还可以通过scRNA数据精确填补未测量的空间分辨多重误差稳健的荧光原位杂交(MERFISH)基因。此外,通过输出OT计划,作者证明了uniPort可以准确地破译小鼠大脑的典型结构,帮助定位乳腺癌区域的三级淋巴结构(TLS),并在基于微阵列的空间数据中揭示癌症异质性。
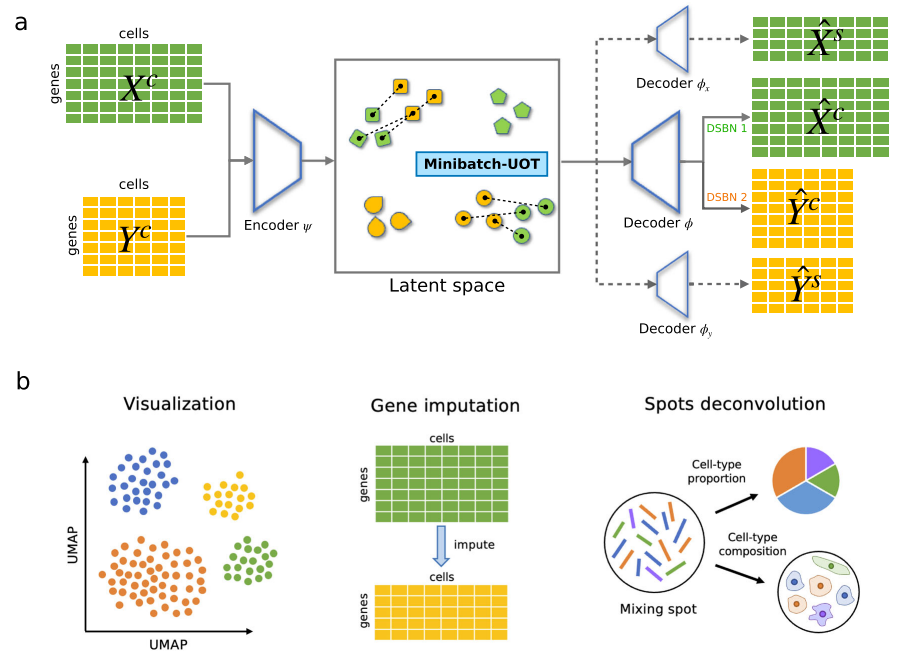
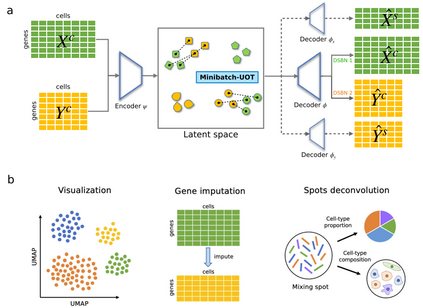
图1 uniPort算法概述
结果
uniPort通过coupled-VAE和Minibatch-UOT嵌入和集成数据集 uniPort在不同模式或技术中采用不同异构单细胞数据集作为输入。uniPort通过耦合变分自编码器(coupled-VAE),利用无数据集编码器将不同数据集的高度可变公共基因集投射到广义细胞嵌入的潜在空间中。然后uniPort重构两个输入项,一个由具有数据集特定批量规范化(DSBN)层的无数据集解码器输入;另一个是通过与每个数据集对应的数据集特定解码器形成的高度可变基因集(图1)。由于一些常见基因在每个数据集中也高度可变,因此在两个输入项之间经常会发现一些重叠的基因。在集成期间,uniPort最大限度地减少了来自不同数据集的潜在空间中细胞嵌入之间的Minibatch-UOT损失。为了获得更好的校准结果,有必要引入损失,特别是当特定数据集的解码器被认为增加了潜在空间中不同数据集的异质性时。同时,小批量策略大大提高了OT的计算效率,使其可扩展到大数据集,而不平衡OT更适合异构数据集成。
作者使用了不同的评估指标来评估uniPort集成单细胞数据的能力。为了量化数据集混合和细胞类型分离,作者使用SCALEX计算了两种评分:批次熵分数用来评估跨数据集混合细胞的程度,轮廓系数用来评估生物学特征的分离。为了对标注聚类的准确性进行基准测试,作者采用了调整兰德指数(ARI)、归一化互信息(NMI)和使用细胞类型标注的F1分数。然后,对于配对数据集,作者使用比真实匹配更接近的样本的平均分数(FOSCTTM)来衡量跨数据集细胞间对应关系的保持情况。
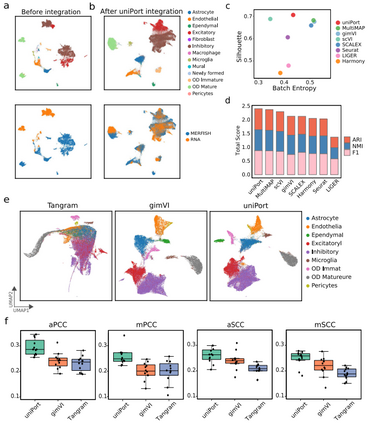
uniPort集成scATAC和scRNA数据 在一个配对的scATAC和scRNA数据集(配对的PBMC数据集)和两个不配对的scATAC和scRNA数据集(基于微流体的PBMC数据集和小鼠脾脏数据集)上,作者将uniPort与目前最先进的单细胞基因组学集成方法进行了基准测试,同时采用一致的流形近似和投影(UMAP)将积分结果进行可视化。
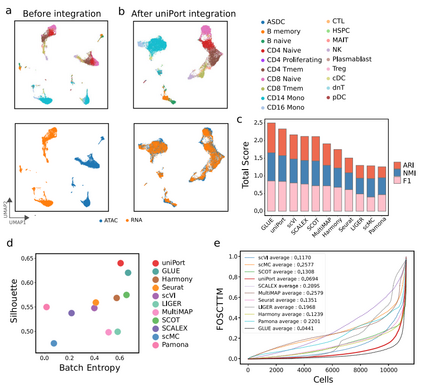
作者首先应用uniPort来集成配对的PBMC数据集(图2),配对信息仅用于性能评估。实验结果表明,uniPort和GLUE的性能最佳,且结果相当(图2c-e)。在所有比较的方法中,uniPort,、Seurat、 Harmony、 SCOT和GLUE在两种模式中准确集成了大多数细胞类型(图2b)。
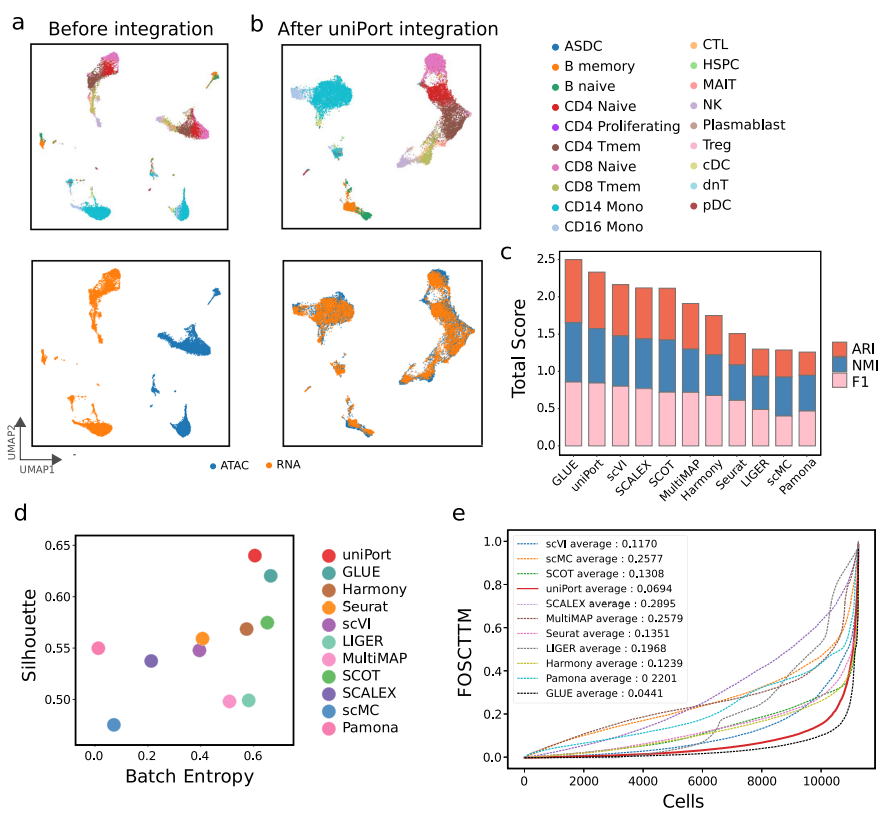
图2 uniPort集成了配对的PBMC数据集
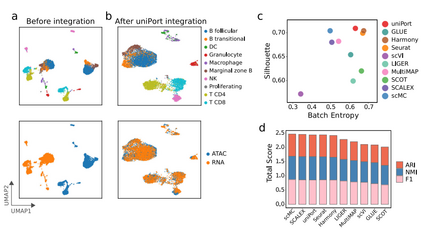
除了集成配对的PBMC数据集,作者还在基于非配对微流体的PBMC数据集上进一步评估了uniPort。结果显示,uniPort准确地集成了scATAC和scRNA数据,并且具有与GLUE、MultiMAP和Harmony相当的竞争性能。此外,作者还在另一个来自小鼠脾脏数据集的未配对的scATAC和scRNA上测试了uniPort (图3)。uniPort、scMC、Harmony和Seurat都取得了较好的性能。总之,在所有方法中,uniPort与最近发表的最先进方法相比表现良好,且在配对和非配对数据集中均显示出准确和稳健的结果。
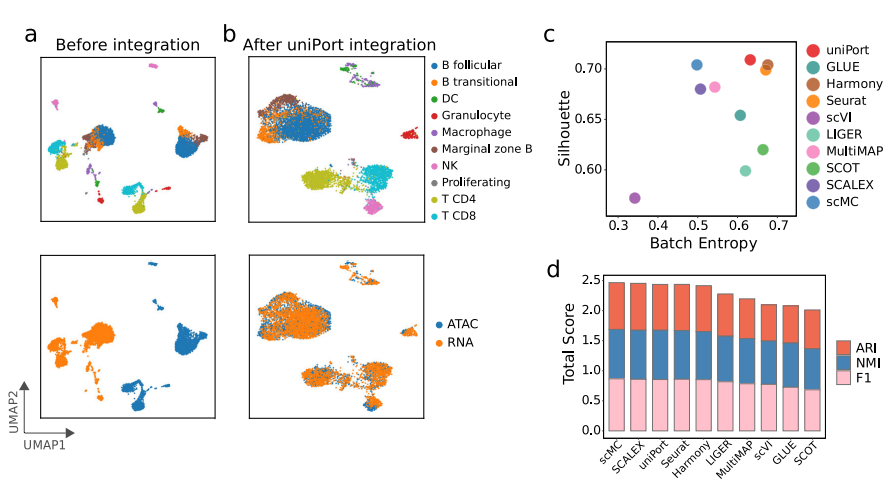
图3 uniPort集成了非配对的小鼠脾脏数据集
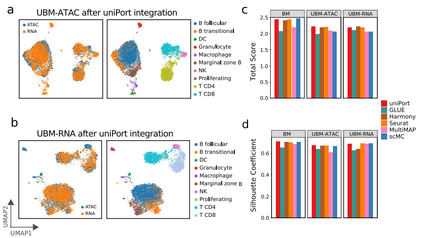
uniPort执行异构数据集的非平衡匹配任务 uniPort最大限度地减少了Minibatch-UOT的损失,适用于不平衡匹配,为异构数据集成提供了强有力的保障。为了评估uniPort在异构数据集成方面的性能,作者分别从小鼠脾脏的scATAC或scRNA中去除一些细胞类型,进行了两项不平衡匹配任务。去除scATAC数据部分类型的集成任务命名为ATAC不平衡匹配(“UBM-ATAC”),将在scRNA数据中删除相同细胞类型的集成任务表示为RNA不平衡匹配(“UBM-RNA”)。同时,为了进行比较,将完整小鼠脾脏数据的集成定义为平衡匹配(“BM”)。
在两种不平衡匹配的情况中,uniPort准确地识别并分离了“DC”、“Granulocyte”、“Macrophage”和“NK”细胞与其他细胞类型,同时很好地对齐了模态共享细胞类型(图4a, b)。将uniPort与GLUE、Harmony、Seurat、MultiMAP和scMC进行比较,可以发现这些方法都在“BM”任务中取得了较高的准确性。但在所有方法中,只有uniPort和Seurat在三种情况下都取得了稳定的性能(图4c, d)。因此,与“BM”的情况相比,当数据集中呈现异质性时,uniPort比其他方法更稳健。
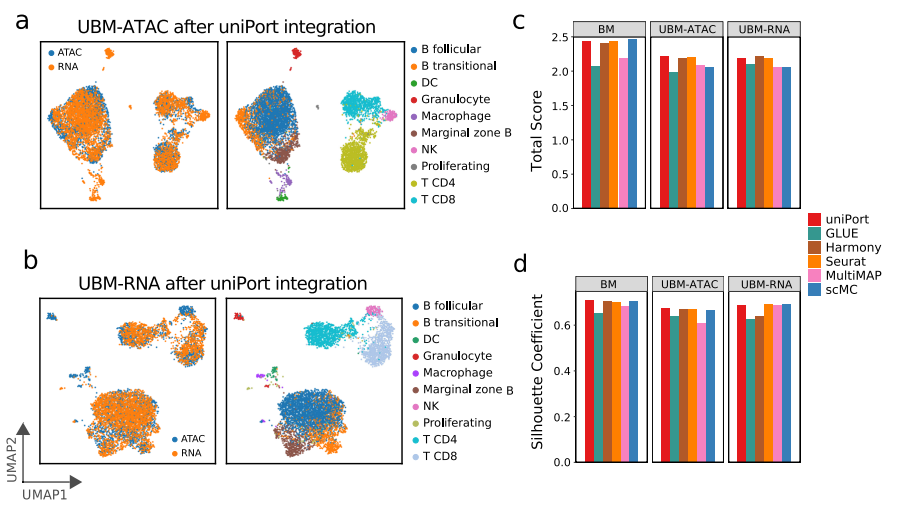
图4 uniPort集成了细胞类型不平衡的小鼠脾脏数据
uniPort集成了MERFISH和scRNA数据 作者进一步考虑了ST和scRNA数据的集成。ST测序技术主要有两种类型:基于高复杂度RNA成像的ST测序技术和基于条形码的ST测序技术。基于高复杂度RNA成像的空间测序具有单细胞精度更高、深度更大的优势,但局限于覆盖率较低的部分测量。为了测试uniPort在基于高复杂度RNA成像数据上的性能,作者应用uniPort集成了MERFISH和scRNA数据。
在MERFISH数据的155个基因中,使用scRNA和MERFISH中的153个共同基因进行集成。应用UMAP将uniPort、Harmony、Seurat、SCALEX、scVI、gimVI和MultiMAP的细胞嵌入结果可视化(图5a,b)。从图中可以看出,uniPort和scVI在鉴别和分离OD 未成熟细胞和其他细胞类型方面的表现优于其他方法。通过再次使用轮廓系数和总分对uniPort与其他方法的集成性能进行基准测试(图5c, d),可发现uniPort优于其他方法。
uniPort为MERFISH数据插补基因 uniPort训练了一个编码器网络,以将跨数据集中具有共同基因的细胞投射到共同细胞嵌入的潜在空间,同时训练了一个解码器网络,以重建具有共同基因和特定基因的细胞。因此,一旦coupled-VAE训练良好,就可以将其视为参考图谱,从而允许uniPort通过图谱根据另一个数据集的共同基因来对一个数据集中的共同基因和特定基因进行插补。插补基因可用于增强空间转录组学的分辨率。
为了探索uniPort的基因插补能力,作者采用gimVI方案从MERFISH的scRNA中插补缺失基因。首先随机选择MERFISH中80%的基因作为训练基因,并保留剩余20%的基因作为测试基因。重复上述步骤12次,得到12个训练和测试基因集。然后用每个训练基因集训练uniPort网络,同时对相应的测试基因集进行插补,并将实验结果与两种最先进的基因插补方法进行比较:gimVI和Tangram。通过应用uniPort、gimVI和Tangram对测试基因进行插补,并使用UMAP对训练和测试基因进行可视化(图5e)。在MERFISH数据集上,与两种比较方法相比,uniPort具有较优的性能 (图5e,f)。
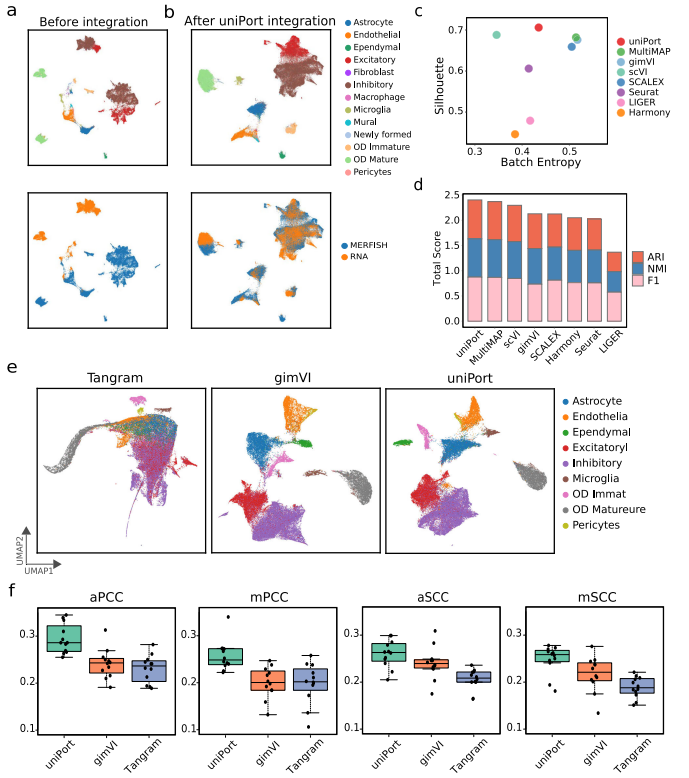
图5 uniPort为MERFISH数据插补
uniPort去卷积合成的STARmap数据 基于条形码的ST更容易获得转录本,覆盖率更高,但仅限于分辨率较低的混合点。接下来,通过将scRNA数据中的标签转移到混合点来实现基于条形码的ST数据的去卷积。uniPort可以提供一个OT计划,该计划代表scRNA和ST数据之间的细胞到混合点的概率对应关系,从而能够根据scRNA数据中的细胞注释来对ST数据的单细胞簇比例进行去卷积。为了评估性能,将uniPort与两种最先进的细胞类型去卷积方法Tangram和SpaOTsc进行了基准测试。实验结果表明,uniPort对比两种方法表现较好。
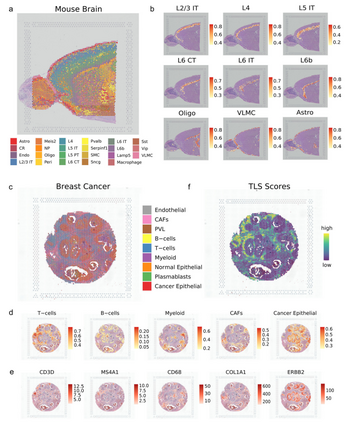
uniPort破译小鼠大脑的典型结构 作者应用uniPort来去卷积现实世界中基于条形码的ST示例。为了估计每个捕获点的细胞类型组成和破译典型的组织结构,作者首先整合了成年小鼠脑ST数据的前切片(10× Visium)。
正如清晰的边界所示,uniPort准确地重建了结构良好的层并去卷积了28种细胞类型(图6a)。多皮质层和区域特异性细胞类型等代表性簇的比例和位置与过往研究高度一致。尽管其解剖结构复杂,但uniPort仍精确地重塑和排列了从边界延伸到中心区域的L2/3L6亚簇 (图6b)。此外,L6层的亚群也被清晰地分离,揭示了该方法对几乎无法察觉的信号的敏感性。因此,无论是标记基因的表达还是大脑的解剖结构,都证明了所提出图谱的稳健性,且可以在基于基因表达的聚类和解剖注释之间建立一致性,并提供比通过视觉检查更彻底和全面的理解。
uniPort帮助定位乳腺癌区域的TLS 癌症的发生和发展通常受到其与异质性肿瘤微环境(TME)的关联影响,而ST可以为其提供生物学见解。为了进一步证明其灵活的效用,作者使用uniPort对 HER2阳性乳腺癌的空间数据进行去卷积,其中包含弥漫性浸润细胞,这使得去卷积位点变得更加困难。如图6c所示,在空间图像上分配了9个主要簇,主要涉及T细胞和癌上皮细胞。此外,实验发现分散在集中富集区域的代表性簇与标记基因表达所显示的区域相一致(图6d,e)。
大量研究表明,免疫细胞浸润的增加与乳腺癌的良好预后高度相关。TLS是近年来在肿瘤或炎症部位发现的一种异位淋巴样器官,被认为是肿瘤患者的预后和预测因素。虽然TLS中存在多种细胞类型,但主要是T细胞和B细胞,它们的联合共定位影响了TME。作者通过对每个点的细胞类型比例进行分析,并通过T细胞和B细胞共定位来识别TLS信号,以使T细胞和B细胞的表达强度相同(图6f)。总体而言,uniPort可以协调不同模态,并满足对组织和疾病代表性结构的高分辨率映射和识别。
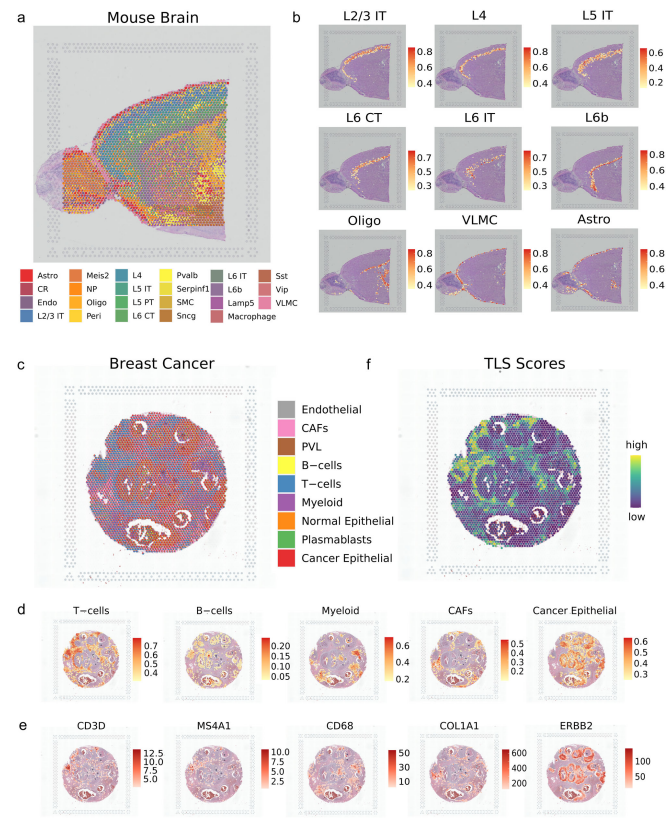
图6 uniPort识别空间转录组学数据中的标志性结构
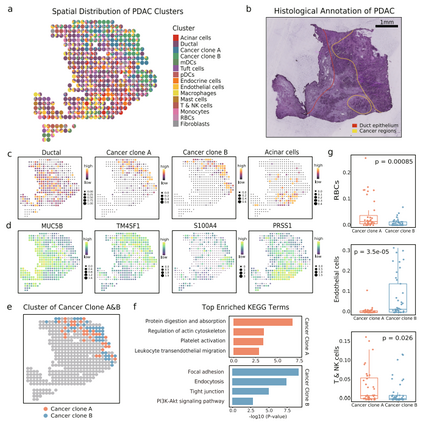
uniPort揭示了基于微阵列的空间数据中的癌症异质性 基于Visium的ST数据区域被限制在每个捕获点直径为55 μm的范围内,这达到了相当于3-30个细胞的中等分辨率。随着位点分辨率的降低,可能会对集成造成潜在的影响,因为成分的混合增长会带来更多的噪声。为了检验uniPort在这种情况下的性能,作者使用基于微阵列的胰腺导管腺癌(PDAC)组织的ST数据进行集成,其直径延伸至100 μm。对1926个单细胞配对的428个位点进行细胞类型去卷积,并分别测量了19736个基因。
通过分解15个主要的簇,可以发现它们表现出正常和肿瘤组成的离散富集和复杂性(图7a)。正常胰腺细胞类型分为导管细胞和腺泡细胞,与以往研究结果一致,保留了与癌细胞明显不同的分布和遗传特征。对于恶性胰腺细胞,根据遗传差异将其分组为癌症克隆A簇和B簇。其次,正常和癌变区域的组织学注释总体上符合其数据驱动的标签(图7b), TME的基本成分由其标记基因表示(图7c, d)。
为了进一步了解癌症亚型的异质性,进一步分析确认它们的身份,以及每个位点所占最大比例(图7e)。KEGG通路将它们分离成不同的功能组件(图7f)。此外,在肿瘤克隆A中,包括红细胞(RBC)、T细胞和自然杀伤(NK)细胞在内的血源性细胞比例显著增加(图7g),这与功能分析的结果一致。综上所述,该方法可以操作不同分辨率的应用光谱,揭示细微的异质性TME。
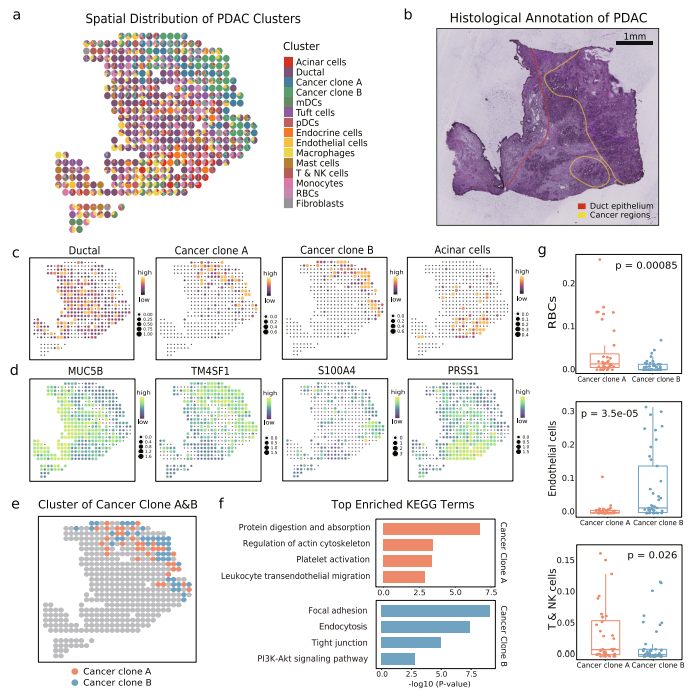
图7 uniPort在基于微阵列的空间数据中识别不同的癌症亚型
总结
本文介绍了一种用于单细胞数据集成的统一深度学习方法uniPort,并将其应用于集成基于转录组学、表观基因组学、空间分辨的高复杂度RNA成像以及条形码的单细胞基因组学。uniPort结合了coupled-VAE和Minibatch-UOT,并利用高度可变的常见基因和数据集特异性基因进行集成。它是一种非线性方法,可将所有数据集投射到一个公共潜在空间,并在数据集之间输出其潜在表示,从而实现可视化和下游分析。
uniPort解决了几个计算方面的挑战,首先是通过使用Minibatch-UOT消除其他基于自编码器模型所需配对细胞的约束。与仅考虑跨数据集常见基因的现有方法不同,作者还利用了每个数据集特有的基因,从而捕获常见基因中不存在的细胞类型异质性。此外,由于coupled-VAE的泛化能力,uniPort通过构建参考图谱显示了其在基因插补方面的能力和潜力。需要指出的是,uniPort甚至可以通过一个数据集中的公共基因来插补另一个数据集中的独特基因,而无需从头开始训练。此外,uniPort还可以输出用于下游分析的OT计划,如灵活的标签迁移学习,用于空间异构数据的去卷积。
在集成大规模异构数据集方面,uniPort具有计算效率和可扩展性,而这对于其他基于OT的方法来说可能是计算上的障碍。目前流行的基于OT的单细胞分析方法是基于全局最优传输,但全局最优传输使得计算非常昂贵。为了解决这一问题,uniPort在基于VAE的单细胞基因组学分析框架中引入了Minibatch-UOT,在每次迭代中只需要求解一个mini-batch传输计划,从而大大降低了计算成本。因此,它可扩展到大数据集。
由于研究中对scATAC的集成是基于基因活性评分,为此作者还测试了uniPort在以不同方法计算基因活性评分时的性能。集成结果显示,uniPort在MAESTRO的基因活性评分上取得了更好的表现,所有的评分都高于Signac,这说明了对基因活性评分建模的重要性。
与最近发表的最先进的方法相比,uniPort始终表现良好,并成功地使用OT计划去卷积空间异构数据。随着配对数据集和各种异质模态的快速发展,本文还通过使用CITE-seq数据和SNARE-seq数据集或没有对齐公共基因的数据集,证明了uniPort对其他类型单细胞数据的普遍适用性。
参考资料
Cao, K., Gong, Q., Hong, Y. et al. A unified computational framework for single-cell data integration with optimal transport. Nat Commun 13, 7419 (2022). https://doi.org/10.1038/s41467-022-35094-8
数据 https://datadryad.org/stash/dataset/doi:10.5061/dryad.8t8s248 https://github.com/liulab-dfci/MAESTRO/blob/master/data/pbmc.ATAC.RData https://github.com/liulab-dfci/MAESTRO/blob/master/data/pbmc.RNA.RData
代码 https://github.com/caokai1073/uniPort