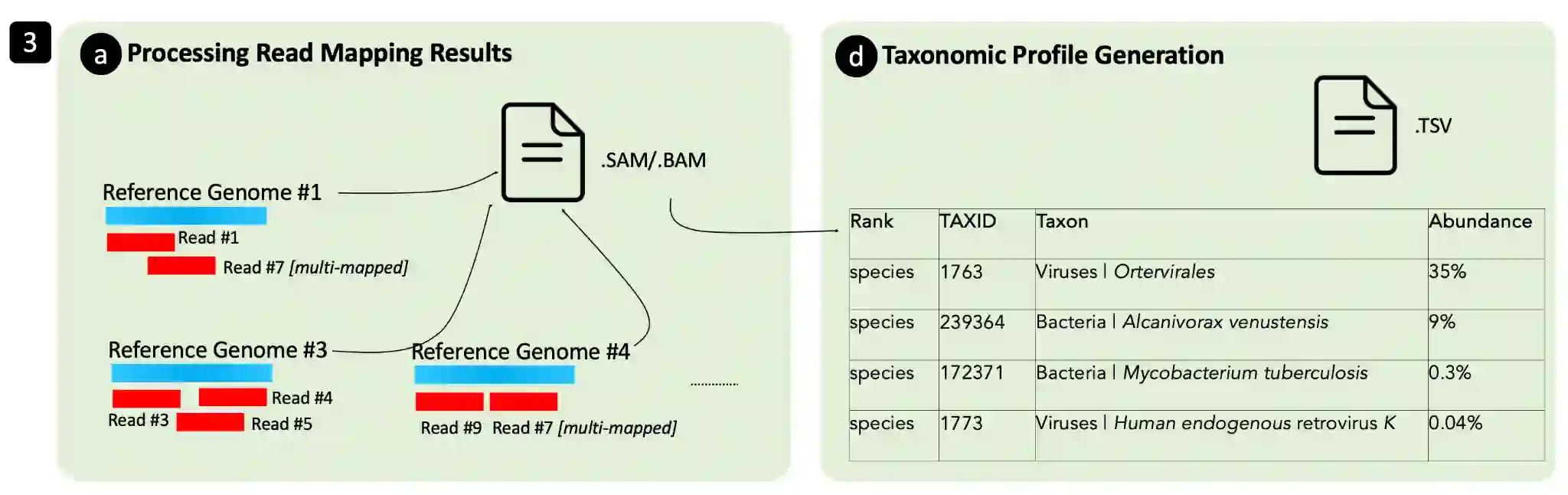
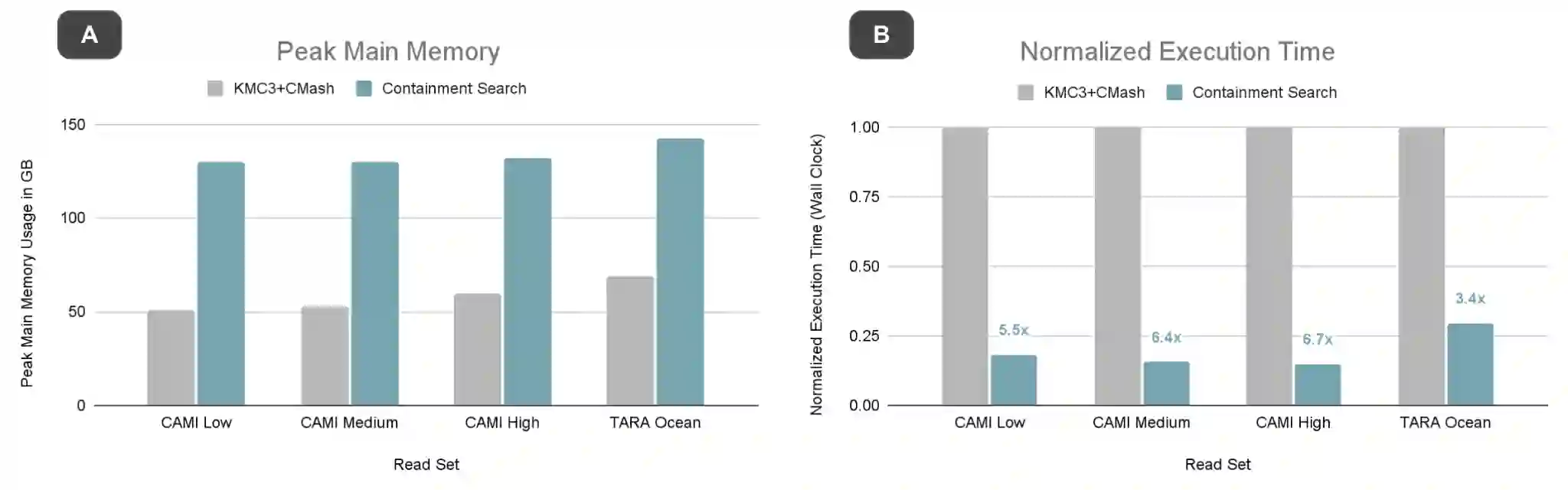
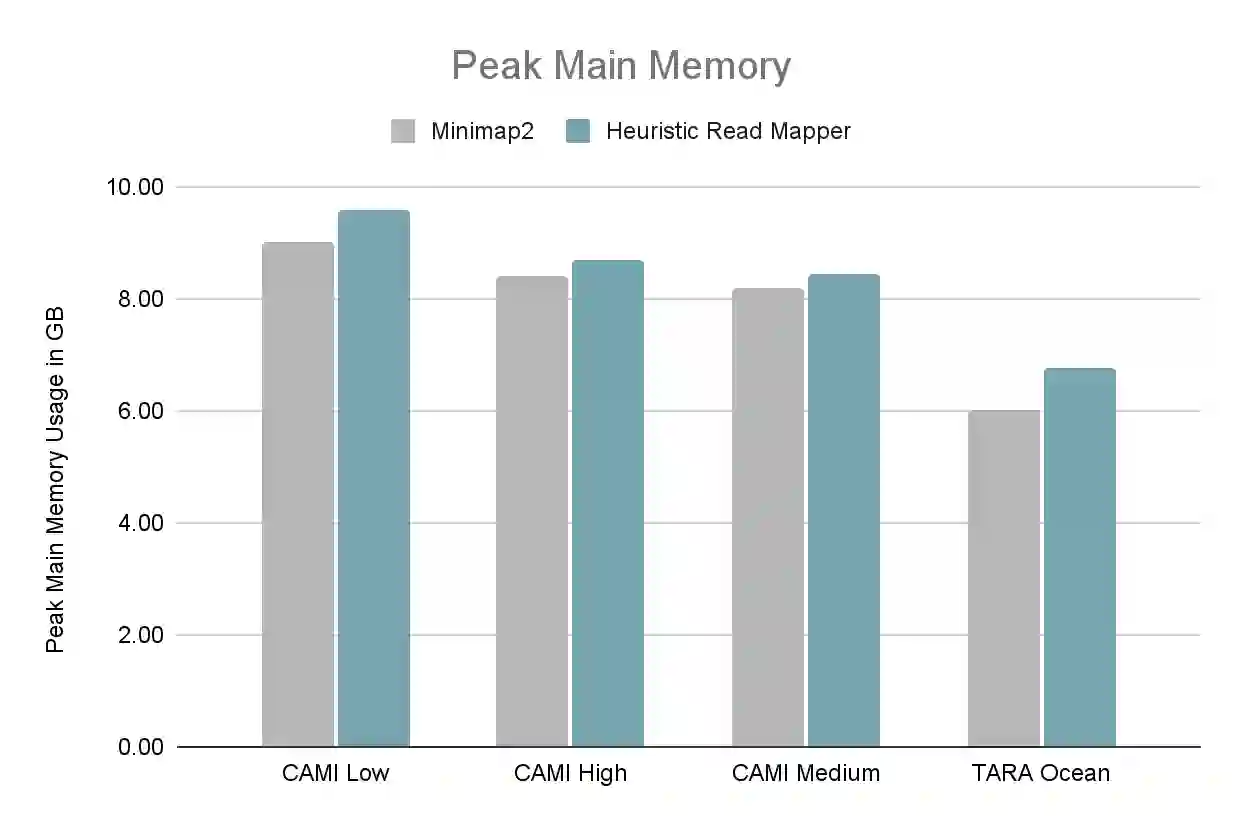
Metagenomics, the study of genome sequences of diverse organisms cohabiting in a shared environment, has experienced significant advancements across various medical and biological fields. Metagenomic analysis is crucial, for instance, in clinical applications such as infectious disease screening and the diagnosis and early detection of diseases such as cancer. A key task in metagenomics is to determine the species present in a sample and their relative abundances. Currently, the field is dominated by either alignment-based tools, which offer high accuracy but are computationally expensive, or alignment-free tools, which are fast but lack the needed accuracy for many applications. In response to this dichotomy, we introduce MetaFast, a tool based on heuristics, to achieve a fundamental improvement in accuracy-runtime tradeoff over existing methods. MetaFast delivers accuracy comparable to the alignment-based and highly accurate tool Metalign but with significantly enhanced efficiency. In MetaFast, we accelerate memory-frugal reference database indexing and filtering. We further employ heuristics to accelerate read mapping. Our evaluation demonstrates that MetaFast achieves a 4x speedup over Metalign without compromising accuracy. MetaFast is publicly available on: https://github.com/CMU-SAFARI/MetaFast.
翻译:暂无翻译