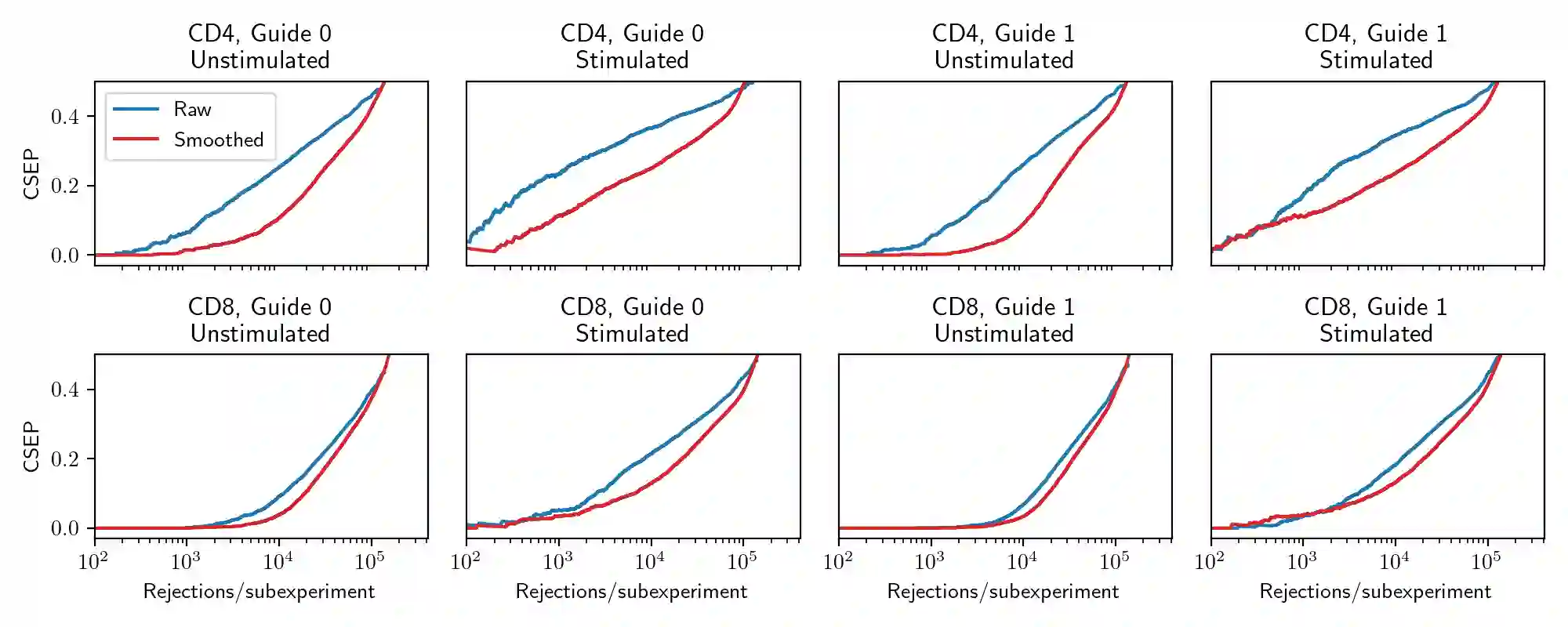
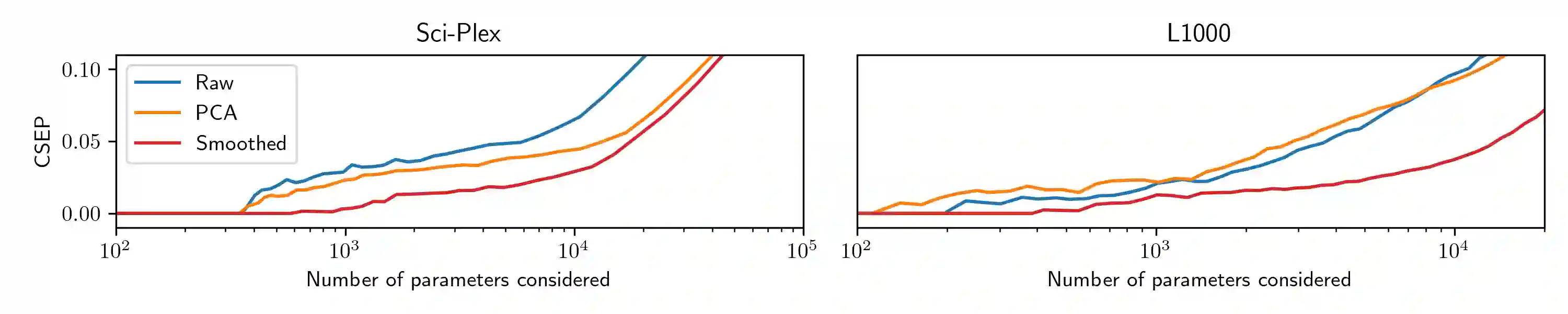
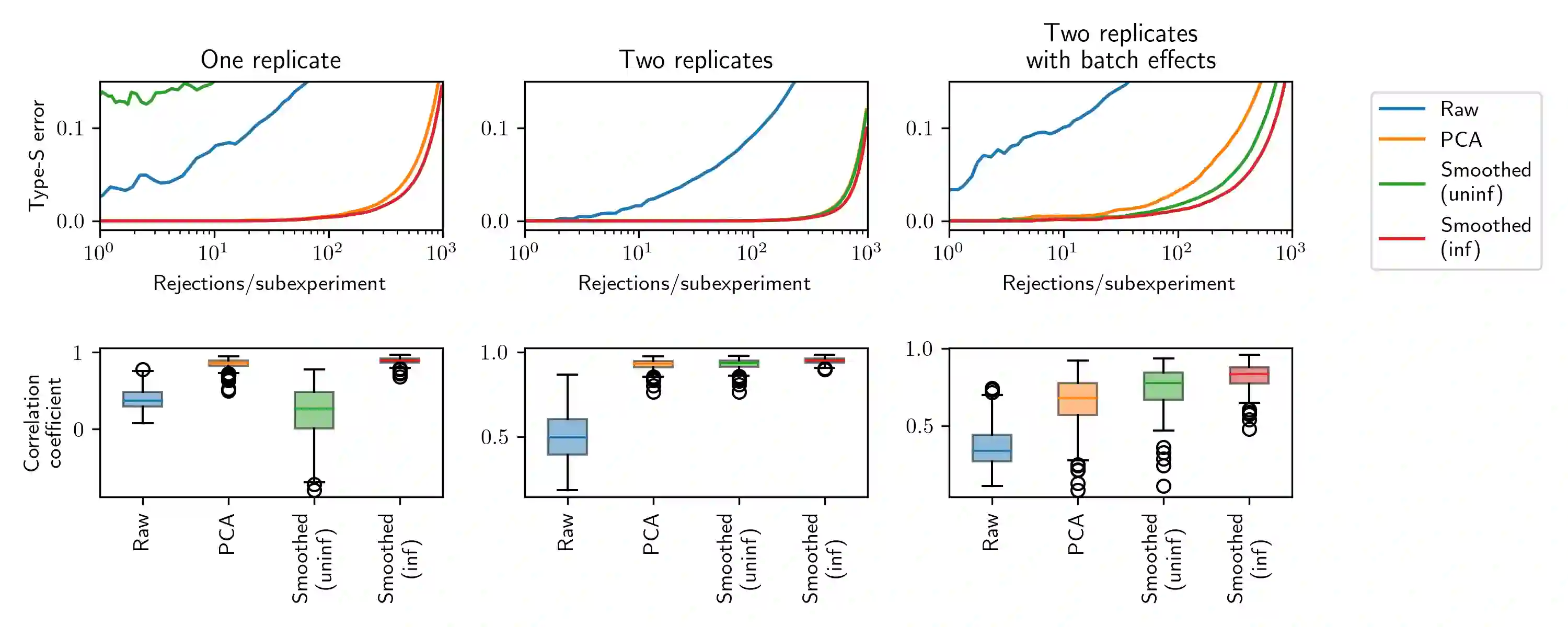
Modern cell-perturbation experiments expose cells to panels of hundreds of stimuli, such as cytokines or CRISPR guides that perform gene knockouts. These experiments are designed to investigate whether a particular gene is upregulated or downregulated by exposure to each treatment. However, due to high levels of experimental noise, typical estimators of whether a gene is up- or down-regulated make many errors. In this paper, we make two contributions. Our first contribution is a new estimator of regulatory effect that makes use of Gaussian processes and factor analysis to leverage auxiliary information about similarities among treatments, such as the chemical similarity among the drugs used to perturb cells. The new estimator typically has lower variance than unregularized estimators, which do not use auxiliary information, but higher bias. To assess whether this new estimator improves accuracy (i.e., achieves a favorable trade-off between bias and variance), we cannot simply compute its error on heldout data as ``ground truth'' about the effects of treatments is unavailable. Our second contribution is a novel data-splitting method to evaluate error rates. This data-splitting method produces valid error bounds using ``sign-valid'' estimators, which by definition have the correct sign more often than not. Using this data-splitting method, through a series of case studies we find that our new estimator, which leverages auxiliary information, can yield a three-fold reduction in type S error rate.
翻译:暂无翻译