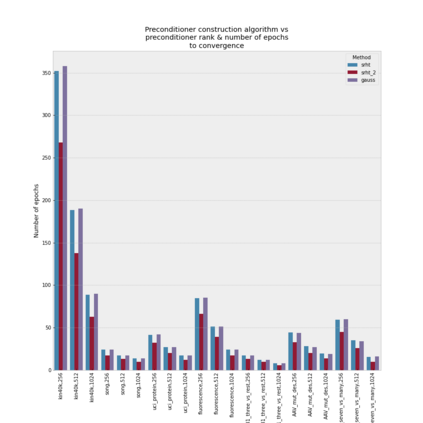
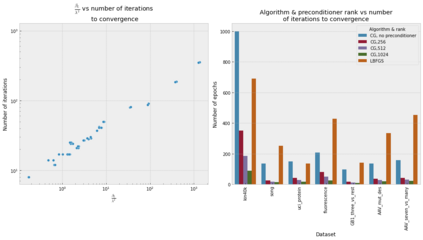
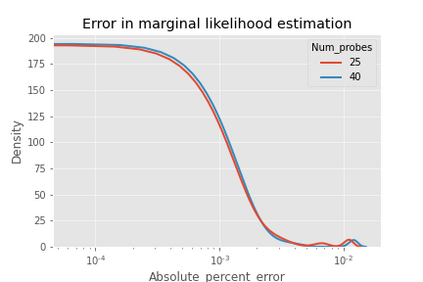
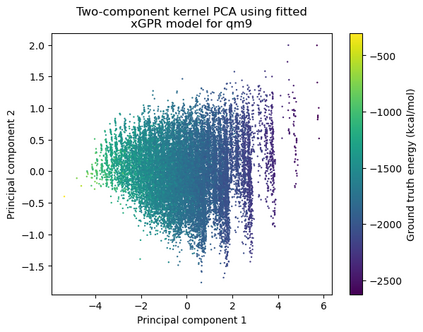
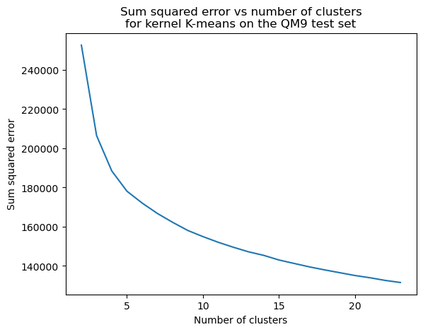
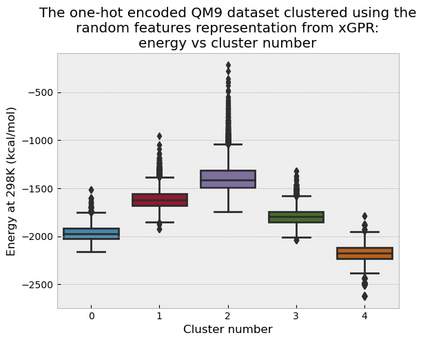
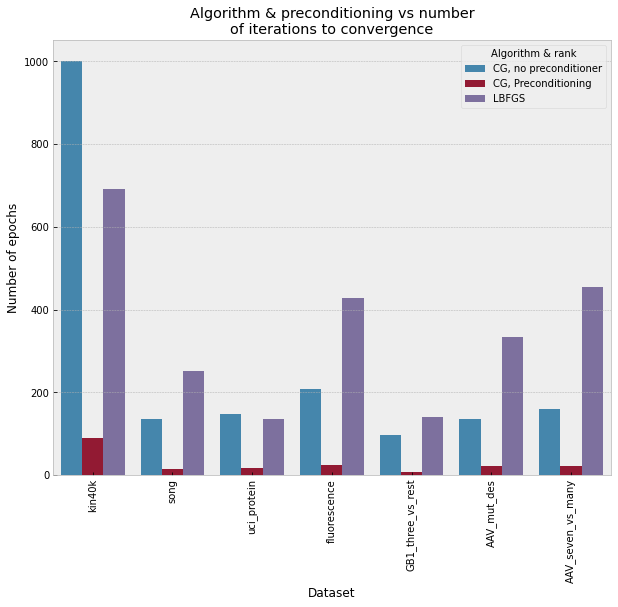
Gaussian process (GP) is a Bayesian model which provides several advantages for regression tasks in machine learning such as reliable quantitation of uncertainty and improved interpretability. Their adoption has been precluded by their excessive computational cost and by the difficulty in adapting them for analyzing sequences (e.g. amino acid and nucleotide sequences) and graphs (e.g. ones representing small molecules). In this study, we develop efficient and scalable approaches for fitting GP models as well as fast convolution kernels which scale linearly with graph or sequence size. We implement these improvements by building an open-source Python library called xGPR. We compare the performance of xGPR with the reported performance of various deep learning models on 20 benchmarks, including small molecule, protein sequence and tabular data. We show that xGRP achieves highly competitive performance with much shorter training time. Furthermore, we also develop new kernels for sequence and graph data and show that xGPR generally outperforms convolutional neural networks on predicting key properties of proteins and small molecules. Importantly, xGPR provides uncertainty information not available from typical deep learning models. Additionally, xGPR provides a representation of the input data that can be used for clustering and data visualization. These results demonstrate that xGPR provides a powerful and generic tool that can be broadly useful in protein engineering and drug discovery.
翻译:暂无翻译