
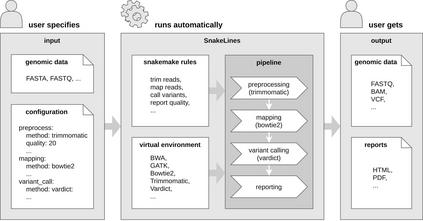
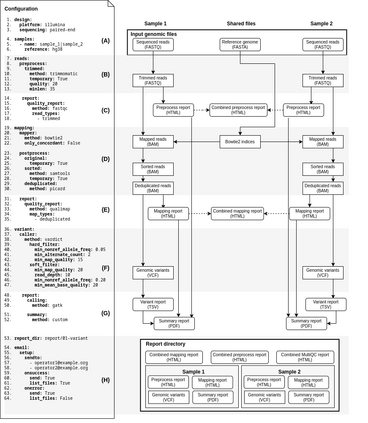
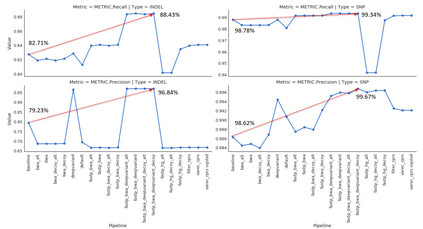
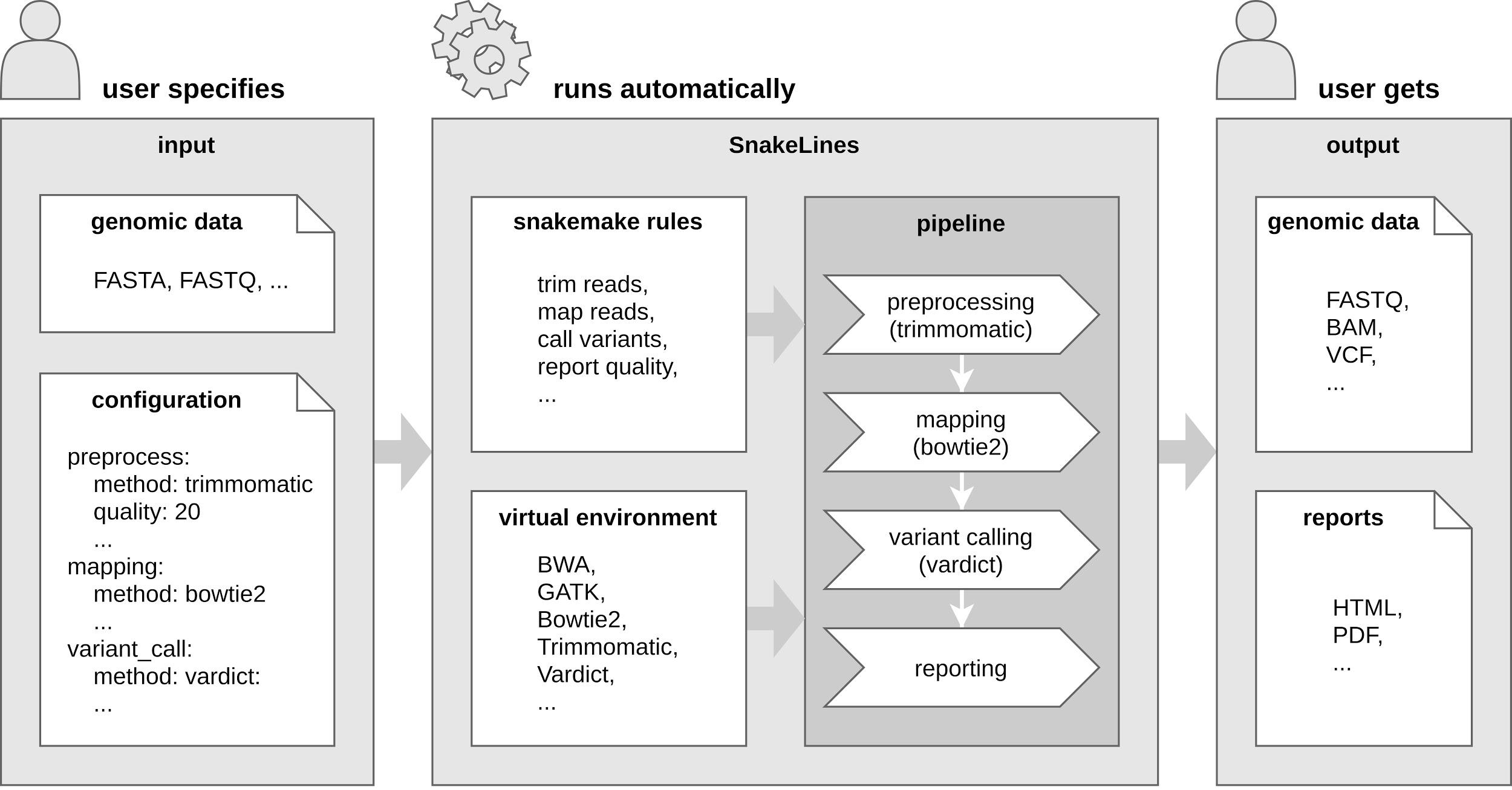
Background: With the rapid growth of massively parallel sequencing technologies, still more laboratories are utilizing sequenced DNA fragments for genomic analyses. Interpretation of sequencing data is, however, strongly dependent on bioinformatics processing, which is often too demanding for clinicians and researchers without a computational background. Another problem represents the reproducibility of computational analyses across separated computational centers with inconsistent versions of installed libraries and bioinformatics tools. Results: We propose an easily extensible set of computational pipelines, called SnakeLines, for processing sequencing reads; including mapping, assembly, variant calling, viral identification, transcriptomics, metagenomics, and methylation analysis. Individual steps of an analysis, along with methods and their parameters can be readily modified in a single configuration file. Provided pipelines are embedded in virtual environments that ensure isolation of required resources from the host operating system, rapid deployment, and reproducibility of analysis across different Unix-based platforms. Conclusion: SnakeLines is a powerful framework for the automation of bioinformatics analyses, with emphasis on a simple set-up, modifications, extensibility, and reproducibility. Keywords: Computational pipeline, framework, massively parallel sequencing, reproducibility, virtual environment
翻译:随着大量平行测序技术的迅速增长,还有更多的实验室正在利用测序DNA碎片进行基因组分析;然而,测序数据的解读在很大程度上取决于生物信息学处理,而生物信息学处理对临床医生和研究人员来说往往要求过高,没有计算背景。另一个问题是,在安装的图书馆和生物信息学工具版本不一致的分离计算中心之间重新复制计算分析。结果:我们提出了一套易于扩展的计算管道,称为 " 蛇线 ",用于处理测序读;包括绘图、组装、变异调用、病毒识别、记录缩写、美格学和甲基化分析。分析的个别步骤,连同方法及其参数,可以很容易地在单一的配置文档中修改。如果管道嵌入虚拟环境中,确保所需资源与主机操作系统隔绝,迅速部署,并复制不同基于Unix平台的分析。结论: " 蛇线 " 是生物信息学分析自动化的强大框架,重点是简单设置、修改、可扩展、可扩展、可扩展、虚拟同步、可复制、虚拟排序框架。
相关内容

- Today (iOS and OS X): widgets for the Today view of Notification Center
- Share (iOS and OS X): post content to web services or share content with others
- Actions (iOS and OS X): app extensions to view or manipulate inside another app
- Photo Editing (iOS): edit a photo or video in Apple's Photos app with extensions from a third-party apps
- Finder Sync (OS X): remote file storage in the Finder with support for Finder content annotation
- Storage Provider (iOS): an interface between files inside an app and other apps on a user's device
- Custom Keyboard (iOS): system-wide alternative keyboards
Source: iOS 8 Extensions: Apple’s Plan for a Powerful App Ecosystem



