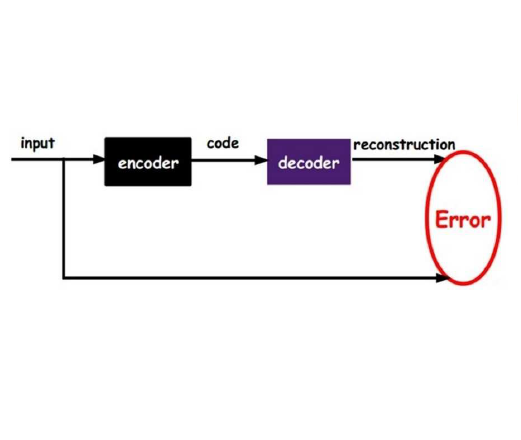
Unsupervised learning on high-dimensional RNA-seq data can reveal molecular subtypes beyond standard labels. We combine an autoencoder-based representation with clustering and stability analysis to search for rare but reproducible genomic subtypes. On the UCI "Gene Expression Cancer RNA-Seq" dataset (801 samples, 20,531 genes; BRCA, COAD, KIRC, LUAD, PRAD), a pan-cancer analysis shows clusters aligning almost perfectly with tissue of origin (Cramer's V = 0.887), serving as a negative control. We therefore reframe the problem within KIRC (n = 146): we select the top 2,000 highly variable genes, standardize them, train a feed-forward autoencoder (128-dimensional latent space), and run k-means for k = 2-10. While global indices favor small k, scanning k with a pre-specified discovery rule (rare < 10 percent and stable with Jaccard >= 0.60 across 20 seeds after Hungarian alignment) yields a simple solution at k = 5 (silhouette = 0.129, DBI = 2.045) with a rare cluster C0 (6.85 percent of patients) that is highly stable (Jaccard = 0.787). Cluster-vs-rest differential expression (Welch's t-test, Benjamini-Hochberg FDR) identifies coherent markers. Overall, pan-cancer clustering is dominated by tissue of origin, whereas a stability-aware within-cancer approach reveals a rare, reproducible KIRC subtype.
翻译:对高维RNA-seq数据进行无监督学习能够揭示超越标准标签的分子亚型。我们结合基于自编码器的表征、聚类与稳定性分析,以搜寻罕见但可复现的基因组亚型。在UCI“基因表达癌症RNA-Seq”数据集(801个样本,20,531个基因;涵盖BRCA、COAD、KIRC、LUAD、PRAD)上进行泛癌分析,结果显示聚类几乎完全与组织来源对齐(Cramer's V = 0.887),这构成了一个阴性对照。因此,我们重新在KIRC(n = 146)内部定义该问题:选取前2,000个高变异基因进行标准化,训练前馈自编码器(128维潜在空间),并对k = 2-10运行k-means聚类。尽管全局指标倾向于较小的k值,但通过预设的发现规则(罕见亚型定义为占比<10%且在20次随机种子下经匈牙利算法对齐后Jaccard指数≥0.60保持稳定)扫描k值,最终在k = 5时获得一个简洁解(轮廓系数 = 0.129,DBI = 2.045),其中包含一个高度稳定的罕见聚类C0(占患者6.85%,Jaccard指数 = 0.787)。通过聚类与其余样本的差异表达分析(Welch's t检验,Benjamini-Hochberg FDR校正)识别出一致性标志物。总体而言,泛癌聚类主要受组织来源主导,而基于稳定性感知的癌种内分析方法则揭示了一种罕见且可复现的KIRC亚型。