
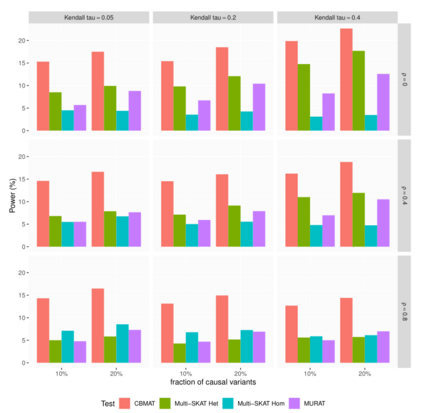
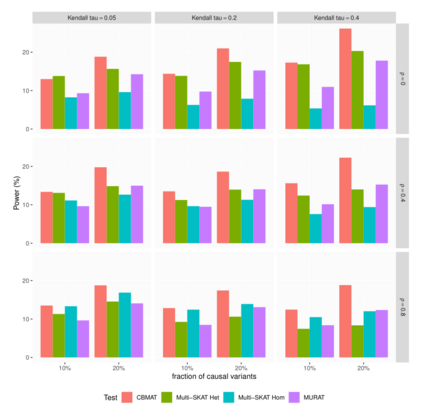
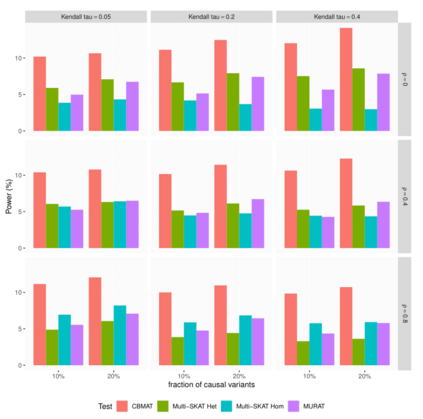
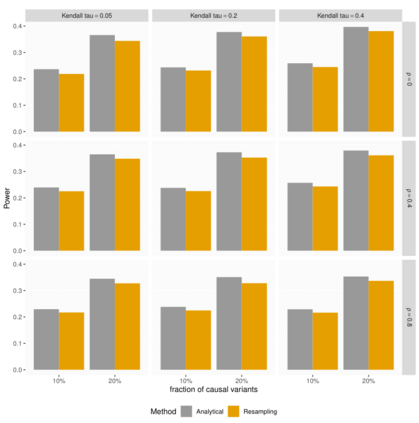
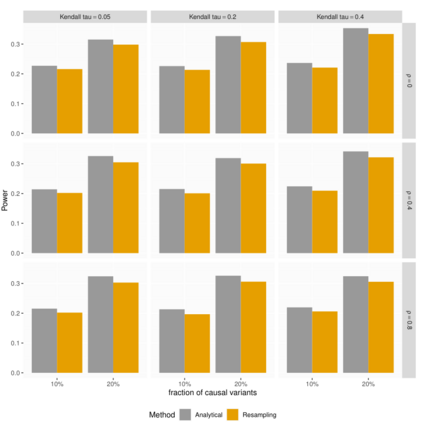
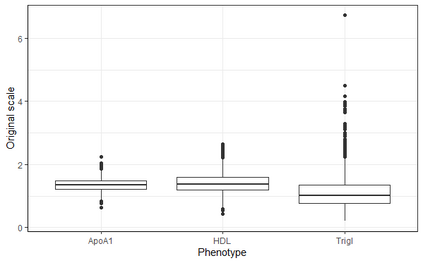
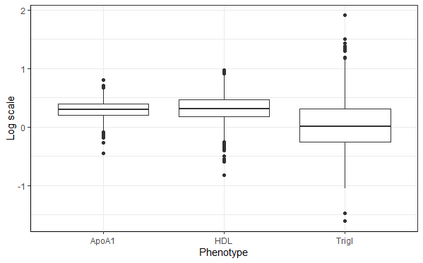
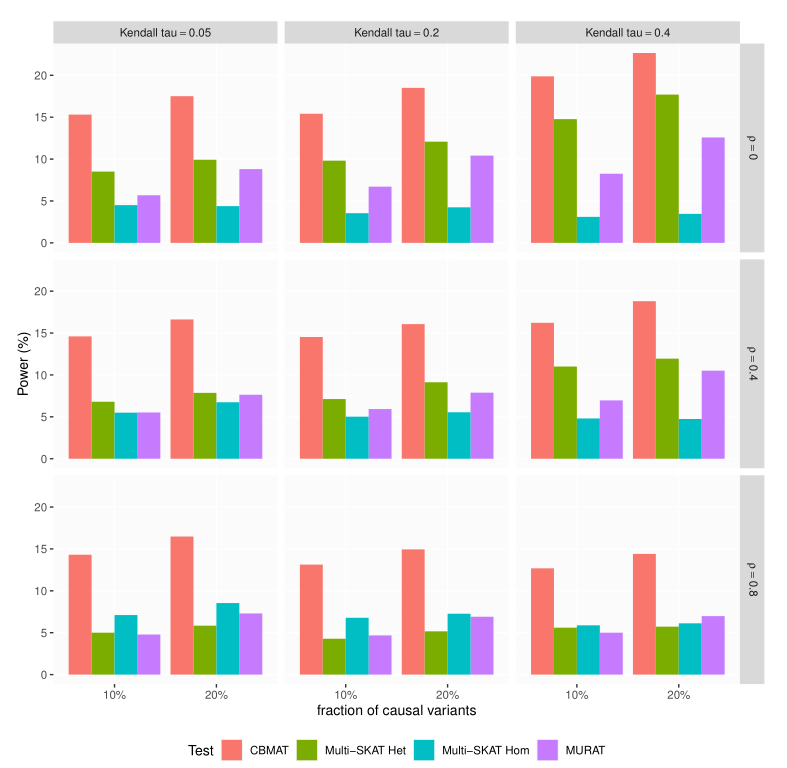
In genome wide association studies (GWAS), researchers are often dealing with non-normally distributed traits or a mixture of discrete-continuous traits. However, most of the current region-based methods rely on multivariate linear mixed models (mvLMMs) and assume a multivariate normal distribution for the phenotypes of interest. Hence, these methods are not applicable to disease or non-normally distributed traits. Therefore, there is a need to develop unified and flexible methods to study association between a set of (possibly rare) genetic variants and non-normal multivariate phenotypes. Copulas are multivariate distribution functions with uniform margins on the $[0, 1]$ interval and they provide suitable models to deal with non-normality of errors in multivariate association studies. We propose a novel unified and flexible Copula-Based Multivariate Association Test (CBMAT) for discovering association between a genetic region and a bivariate continuous or mixed phenotype. We also derive a data-driven analytic p-value procedure of the proposed region-based score-type test. Through simulation studies, we demonstrate that CBMAT has well controlled type I error rates and higher power to detect associations compared with other existing methods, for discrete and non-normally distributed traits. At last, we apply CBMAT to detect the association between two genes located on chromosome 11 and several lipid levels measured on 1,477 subjects from the ASLPAC study.
翻译:在基因组广泛联系研究(GWAS)中,研究人员往往处理非正常分布的遗传变异或混合的多变性特征,但是,目前基于区域的方法大多依赖多变线性混合模型(mvLMMs),对感兴趣的同系物类型采取多变的正常分配办法,因此,这些方法不适用于疾病或非正常分布的特性,因此,有必要制定统一和灵活的方法,研究一组(可能很少见的)遗传变异体和非常态多变异体型(多变异体型)和非常态多变性多变性苯型。 Copulas是多种变异性分布功能,在$[10,1]的间隔上具有统一的差值,它们为处理多种变异性联系研究中的错误的不常态性正常分布提供了适当的模式。因此,我们建议采用新的统一和灵活的CUBAT-多变性协会测试(CBAT),以发现基因区域和双变异性连续或混合的同系型型。我们还从拟议的C-AAT型不同测算的测算和直系物型型的内测算方法,以现有的C-C-SDral-ral-ration-ration-C型测算法测试了现有不同等级等级等级等级等级的两种测算方法。
相关内容

Source: Apple - iOS 8

