npj: 基于结构描述符机器学习—纳米团簇表面的氢吸附能
海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

纳米团簇表面析氢反应的催化活性取决于氢吸附位点的结构,预测团簇的催化活性需要对各种可能的吸附结构开展模拟,计算量庞大。采用基于描述符的机器学习可以显著降低相关预测的工作量。
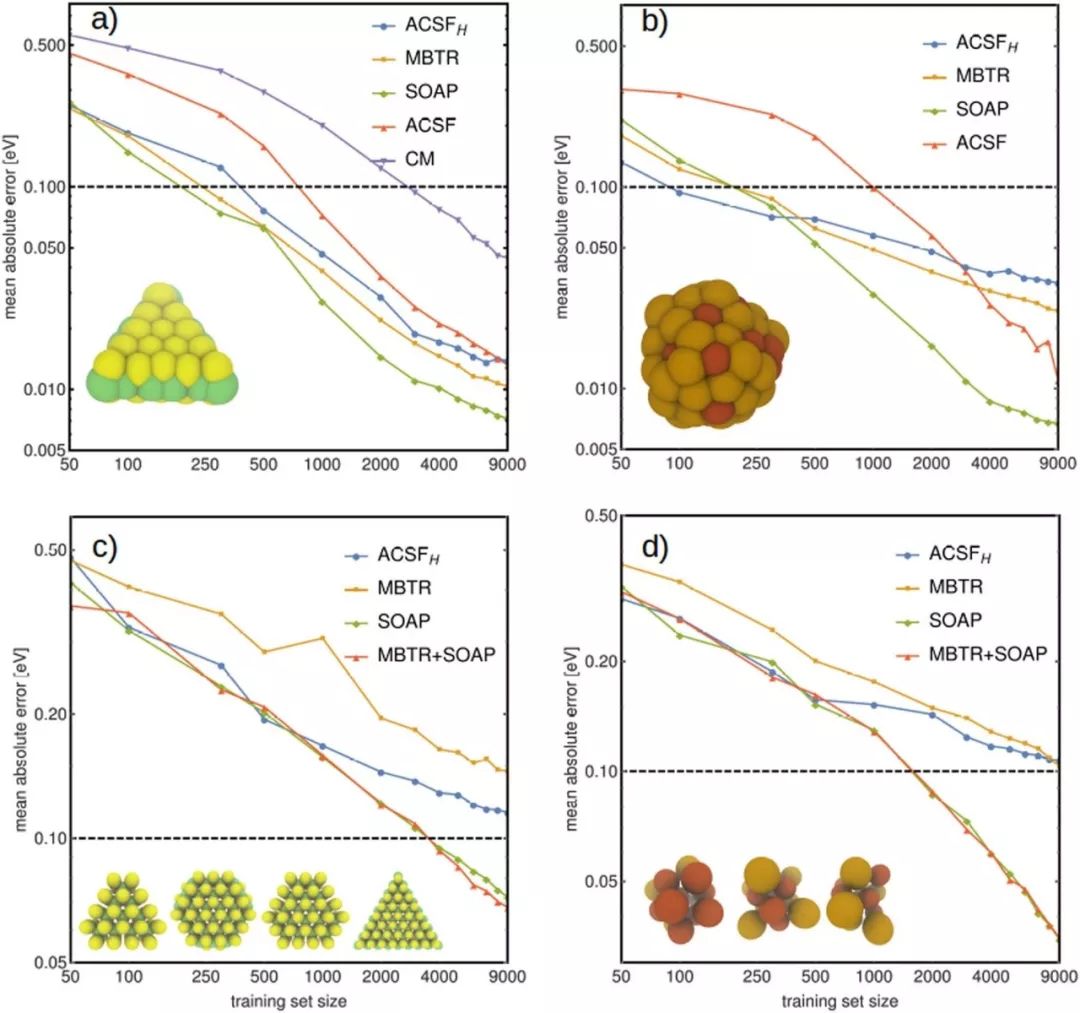
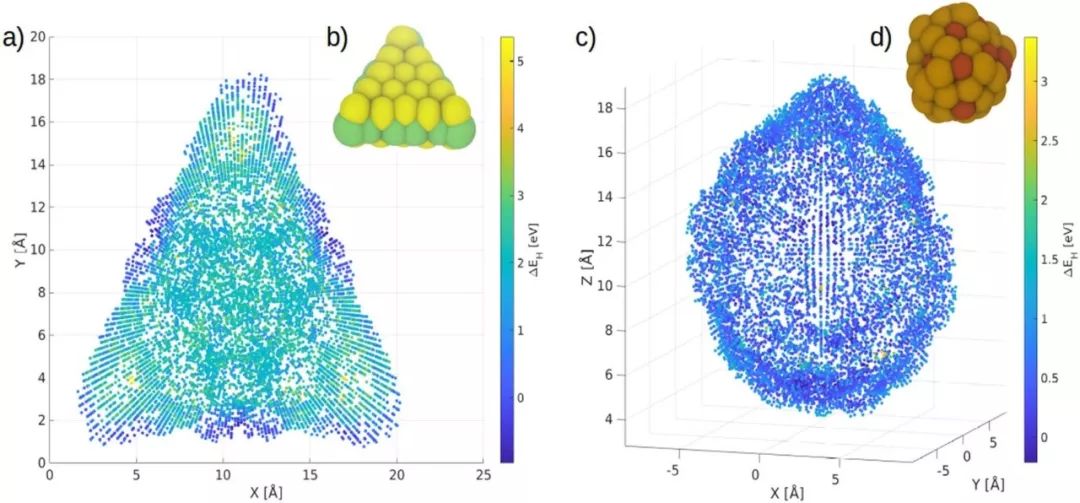
来自芬兰阿尔托大学的Adam Foster教授等分析了目前最先进的结构描述符,即原子位置平滑重叠(SOAP)、多体张量表示(MBTR)和原子中心对称函数(ACSF)用于机器学习纳米团簇表面氢吸附自由能的可靠性。他们以2D的MoS2和AuxCuy合金作为测试体系,扫描了纳米团簇表面氢吸附的势能面,比较了不同描述符用于核岭回归的预测性能。通过对91个MoS 2纳米团簇和24个AuxCuy纳米团簇组成的数据集的分析表明,SOAP相比其他描述符具有显著优越的预测性能,可以作为吸附能预测的候选工具。
该文近期发表于npjComputational Materials 4:3 (2018),英文标题与摘要如下,点击左下角“阅读原文”可以自由获取论文PDF。
Machine learning hydrogen adsorption on nanoclusters through structural descriptors
Marc O. J. Jäger, Eiaki V. Morooka, Filippo Federici Canova, Lauri Himanen & Adam S. Foster
Catalytic activity of the hydrogen evolution reaction on nanoclusters depends on diverse adsorption site structures. Machine learning reduces the cost for modelling those sites with the aid of descriptors. We analysed the performance of state-of-the-art structural descriptors Smooth Overlap of Atomic Positions, Many-Body Tensor Representation and Atom-Centered Symmetry Functions while predicting the hydrogen adsorption (free) energy on the surface of nanoclusters. The 2D-material molybdenum disulphide and the alloy copper–gold functioned as test systems. Potential energy scans of hydrogen on the cluster surfaces were conducted to compare the accuracy of the descriptors in kernel ridge regression. By having recourse to data sets of 91 molybdenum disulphide clusters and 24 copper–gold clusters, we found that the mean absolute error could be reduced by machine learning on different clusters simultaneously rather than separately. The adsorption energy was explained by the local descriptor Smooth Overlap of Atomic Positions, combining it with the global descriptor Many-Body Tensor Representation did not improve the overall accuracy. We concluded that fitting of potential energy surfaces could be reduced significantly by merging data from different nanoclusters.
扩展阅读
本文系网易新闻·网易号“各有态度”特色内容
媒体转载联系授权请看下方


