IJMS|抗体结构建模方法综述
近日,IJMS发表了题为"VHH Structural Modelling Approaches:A Critical Review"的抗体结构建模方法的综述,介绍了应用于给定抗体Abs、VHH(骆驼单链抗体的VH 结构域)序列的 3D 结构建模方法的最广泛的概述(共 21 个)。除了对抗体建模历史的概述之外,作者展示了软件是如何构建Abs、VHH的结构预测,并给出了例子。

Abs和VHH特异性
Abs和VHH具有特定的全局和局部拓扑,即FRs和DCRs或VH/VL界面。因此已经开发了一些特定的Abs建模工具。当然,经典的蛋白质建模工具(下图a)可用于Abs和VHH。由于Abs在生物技术和生物医学领域的各种影响及其多样性。因此出现了对专用Abs工具的需求(下图b)。
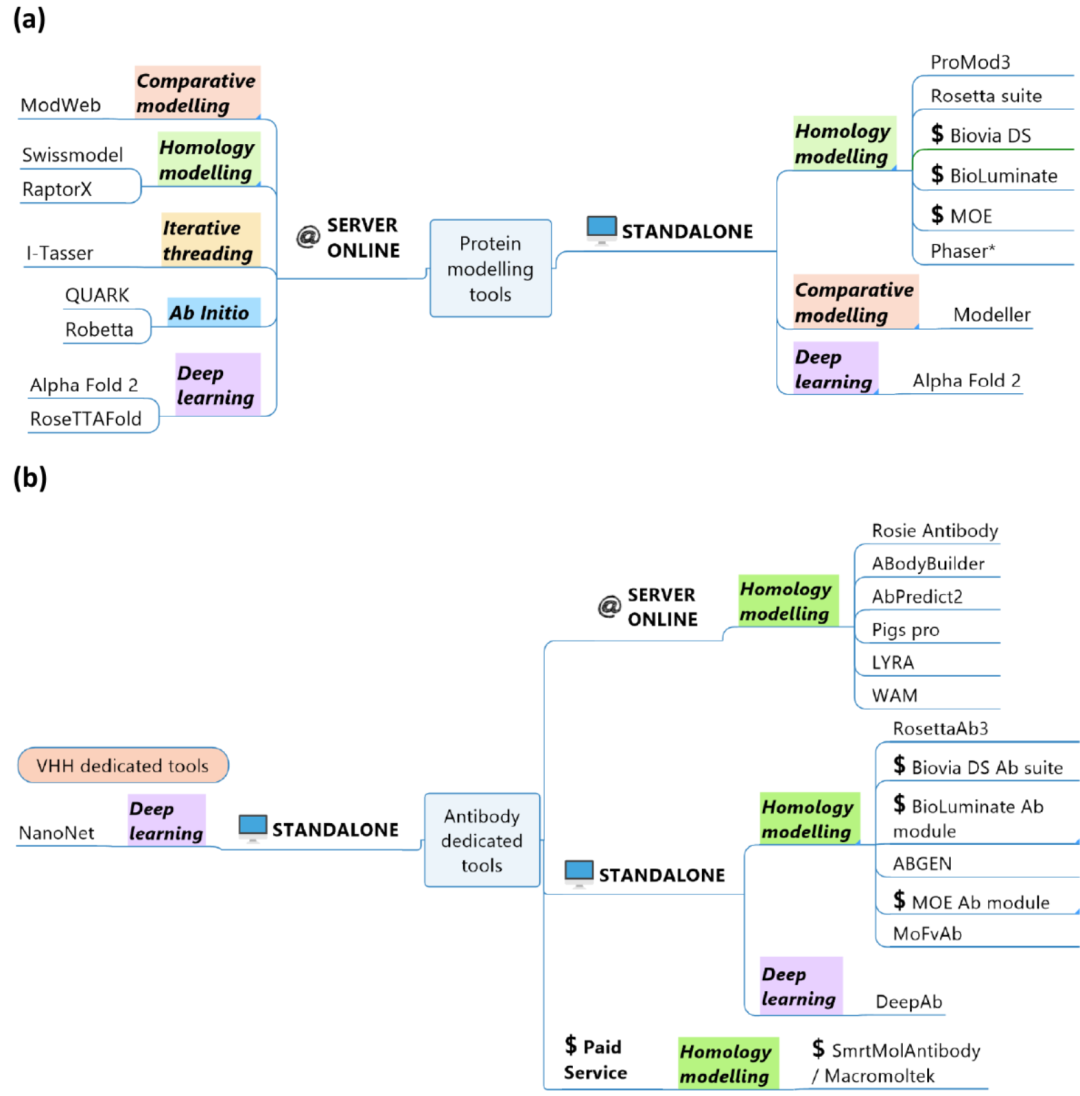
图:蛋白质结构预测工具。(a) 通用蛋白质建模工具和 (b)抗体和VHH建模工具总结。所有工具都根据其可用性(独立或在线服务器、学术或商业)和使用方法(比较/同源建模、threading、从头设计和深度学习)
01 Modeller和ModWeb
Modeller 可能是最重要的比较/同源建模软件。它于1993年开发,目前仍在维护(https://salilab.org/modeller/,最后访问日期:2022 年2月2日)。Modeller 自动生成一个包含所有非氢原子的模型,该模型基于(多个)序列比对,用于使用已知相关结构建模的序列(由用户提供)。它将带有一个或多个结构模板序列的序列查询的比对文件作为输入,存在不同的细化选项。经过多个循环的构建和模型评估后,生成了选定数量的结构模型。用户必须选择与最低 DOPE 分数相关联的最佳分数。此外,开发人员提出了一个名为 ModWeb 的在线 Web 服务器,它使用PSI-BLAST和IMPALA来寻找兼容的结构模板和 Modeller 来构建结构模型 。
02 ABGEN
ABGEN 软件于 1996 年问世。ABGEN 是一种基于同源性的算法,可对抗体进行建模,与 Modeller 有一些很强的相似性。它由称为 ABalign 和 ABbuild 的两个模块组成。ABalign 为重链和轻链找到模板,并为每个识别的模板提供一致性分数。ABbuild 基于选择的最佳模板,为所有链构建刚性模型;然后,优化loops和侧链。最后,ABGEN 包括使用 XPLOR和 GROMOS的专用动力学方法来改进获得的模型。ABGEN 是第一个专门优化 CDR loops的。
03 Web Antibody Modelling
Web Antibody Modelling是一种非常有趣的分层方法,发表于 2000,专门用于抗体建模。用五个连续的步骤来描述:
a) 执行相似性搜索以构建框架(主链和侧链)和具有更接近结构的传统loops backbones(就序列相似性而言)。
b) 使用CONGEN,传统的环侧链是使用迭代有放回技术构建的,以搜索全局最小能量构象。
c) 根据环长度,通过再次直接使用 PDB 或 CONGEN 来产生替代构象。
d) 测试了特定的solvent-modified的共价力场以评估不同的构象。
e) 最后,从五个最低能量构象中选择构象。最终模型是一组扭转角最接近标准扭转角的构象。
04 SWISS-MODEL
SWISS-MODEL 是著名的在线蛋白质同源性建模服务器,于 2003 年首次发布.其基础算法来自于 Modeller 算法的原理(见上文)。compatibel sequences的研究由 PSI-BLAST 结合精细的 HHblits进行。然后,用户可以选择要使用的模板。一个有趣的点是为评估所构建的几个模型的质量而提供了不同方法,即全局模型质量估计(GMQE)。结构模型的构建是使用本地开发的 OpenStructure 完成的,即用于计算结构生物学的集成软件框架。以类似的方式,他们还开发了评估模型质量的方法,即 QMEAN 评分函数。虽然它是一种通用方法,但它已被用于不同经典抗体的建模。
05 MoFvAb
MoFvAb 发表于2015 年,重点是“对抗体的 Fv 区域进行建模”。第一步,它使用 WolfGuy 编号系统注释 FR 和 CDR。基于这些片段,识别出重链和轻链的模板。除 CDR-H3 外,所有链均产生刚性模型。后者经历从头构建。下一步,CDR relaxed,并基于邻域算法进行侧链细化。最后的步骤涉及 VH/VL 方向预测和细化、旋转异构体优化和 CHARMM 最小化。该方法在 AMA-I 和 -II 数据集上进行了测试,结果良好。
06 免疫球蛋白结构预测
这些网络服务器是由 Anna Tramontano 的小组开发的。Anna Tramontano之前曾使用经典方法研究抗体结构、序列-结构关系和建模。第一个版本名为“Prediction of ImmunoGlobulin Structure” (PIGS) 于 2008 年发表,因为作者发现 WAM 有许多局限性并且不是很灵活。根据先前研究的结果,CDRH3 根据其长度进行了不同的建模 。PIGS 为用户提供的序列提出了复杂 VH/VL 的结构模型。这些方法的评估在原始论文中进行了测试,也在 AMA-I 和-II 比赛中进行了测试,取得了良好的结果。第二个版本名为 PIGS PRO,于 2017 年提出。作为WAM,它可以分为五个步骤:
a) 准备框架:使用与来自 PDB 的蛋白质结构的序列同一性来选择框架的结构模板。
b) 构建了六个 CDR 中的五个:CDR L1-L3 、 H1 和 H2 是通过从具有相同传统结构的抗体中获取构象而丢弃一致序列来建模的。
c) 为了完成,使用与查询序列具有最高序列同一性的结构模板提出了CDRH3 loop。
d) 全部合并:对复杂的 VH-VL 进行建模。
e) 最后,SCWRL 用于优化预测模型的侧链构象。
07 AbodyBuilder
ABodyBuilder 是 Deane 实验室开发的用于抗体结构预测的专用工具。该算法首先注释并找到单独的 VH 和 VL 以及复杂的 VH-VL的模板。正如经常看到的那样,该方法是分层的。FREAD 算法尝试识别 CDR 的模板。如果没有找到模板,则从头开始触发 Sphinx 循环。最后一步是使用 IMGT 位置依赖性旋转异构体库,用 PEARS 预测侧链构象。一个有趣的特征是使用所得抗体模型的置信值估计预期 RMSD。作为一个有价值的例子,该方法最近已被用于分析 SARS-CoV-1 特异性 V H H,以了解 CDR 区域的构象多样性、亲和力和稳定性 。
08 淋巴细胞受体自动化建模
淋巴细胞受体自动化建模 (LYRA) 是一种改进的方法来预测 B 细胞和 T 细胞受体结构模型,作为在线网络服务器 ( https://services.healthtech.dtu.dk/service.php?LYRA ) -1.0,上次访问日期:2022 年 2 月 8 日)作者在序列和结构方面都取得了重要进展。这些序列已经用 Abhinandan 和 Martin采用的 Kabat-Chothia 方法进行了注释和编号,并减少了冗余。使用隐马尔可夫模型完成了compatible最优序列的研究。最佳评分曲线用于推断受体(TCR 或 BCR)和链类型(重链或轻链)。在这个阶段,比对被重新认证以正确识别重链和轻链。重要的一点是通过查看(大部分)20 个不同的结构模板来确保不存在碰撞。类似地,为了改进它,基于特定 CDR 聚类在定义的库上搜索 CDR。最后,为了创建最终模型,组装两条链的模板,并重新打包侧链。这个工作过程很快,平均不到一分钟。
09 Phyre2
Phyre及其继任者 Phyre2是与 SWISS-MODEL 等技术非常相似的通用方法,它们将进化信息与蛋白质结构数据集的专门分析相结合。它是一个可用的在线网络服务器(http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index,最后访问日期:2022 年 2 月 9 日)。Phyre2 有两种模式:正常模式和强化模式。
正常模式包括三个步骤:
a) HHblits和HHsearch检测相关序列(有或没有结构)以产生多序列比对并为输入序列搜索足够的模板。一旦确定了模板,就可以构建模型,但大致只使用backbones。
b) 执行一个特定步骤对其中发现这些模型中的插入缺失(插入和删除)来进行优化loop建模。
c) 侧链被嫁接以构建最终的 Phyre2 模型。
10 RaptorX
RaptorX 是一个自动化服务器,它执行基于模板的三级结构建模。它源自 RAPTOR,一种经典的threading方法。RaptorX 与 RAPTOR 相比的改进在于使用具有条件随机场 (CRF) 的非线性对齐评分函数,提供局部熵。第一步,将序列切割成域,进行不同特征的预测,例如无序预测。然后,通过线程过程搜索和排列模板。对齐质量由人工神经网络评估。最后,RaptorX 提供了多个最终模型,包括基于多个排名靠前的模板生成的模型。最新版本的 RaptorX 现在已经进行了改进,包括一个深度卷积神经域 (DeepCNF) 来预测二级结构。DeepCNF 有两个组件:深度卷积神经网络 (DCNN) 和条件随机场 (CRF),用于作为 CRF 的输入和标签(输出)层。它最近通过使用深度学习来预测蛋白质接触得到了丰富。
11 Rosetta,Robetta,RosettaAntibody
在David Baker的指导下发展起来的Rostta故事始于上个世纪。最初,它主要包括从头开始的方法,但逐渐地,它演变为一种从头策略,该策略结合了从数据集中的序列搜索中获得的小结构片段。添加进化信息和局部蛋白质约束提高了结果的质量。最后一轮的各种改进导致删除了non-native contacts。最后,替代结构模型的intensive production和来自同源序列的模型聚类有助于使 Rosetta 在结构模型的命题中成为一个非常成功的工具。Rosetta 的发展令人印象深刻,可以通过不同的 prims 进行分析。RosettaCM 致力于优化比较建模步骤,与最好的类似方法竞争。一个名为 Robetta 的网络服务器是免费提供的(https://robetta.bakerlab.org/,最后访问日期:2022 年 2 月 10 日)。
从全面的Rosetta 软件中,完成了一种特异性抗体开发,即 RosettaAntibody 。该工具可在线获取,并可通过 Rosie 使用(https://rosie.rosettacommons.org/antibody/,最后访问日期:2022 年 2 月 10 日)。该协议由给定抗体序列的三个步骤组成:
a) 在初始步骤中,使用 Kabat CDR 定义识别 CDR,并使用 Chothia 方案重新编号残基。然后对所有框架和六个 CDR 中的五个(CDRH1 和 CDRH2 和 CDRL1-CDR3)进行模板选择,
b) 从选定的模板中,使用同源建模创建初步模型,因为它被证明比完全从头方法更准确。
c) CDRH3 从头循环建模完成了模型预测,同时优化了 VH-VL 界面。
由于 Rosetta 建模套件还采用了对接方法,因此最后一种针对抗体进行了优化。然而,RosettaAntibody 对于单链抗体的效率似乎较低,因此对于 V H Hs 也是如此。因此,对loop定义进行了特定调整以更好地考虑 V H H CDR,尤其是 CDR3的特异性。
12 AbPredict2
AbPredict 通过两个版本在 Rosetta 建模套件中实现:ABPredict1和 ABPredict2。它可以下载(http://abpredict.weizmann.ac.il/bin/steps,最后访问日期:2022 年 2 月 14 日)。简而言之,AbPredict 利用已知的抗体 3D 结构,并将主链构象与低能蒙特卡罗搜索完美结合。
AbPredict 方法基于五个独立的数据库,包括 VH、VL、CDRH3、CDRL3 片段的骨架扭转角以及重链和轻链的刚体方向。初始化通过组合来自五个数据库的随机段开始。执行有偏差的模拟退火蒙特卡罗 (SA-MC) 采样,具有数千条独立的轨迹。从这些轨迹中的每一个种提取最低能量构象。从后面的构象列表中,最终预测的抗体模型是具有最低能量的模型。
AbPredict 已使用 AMA-II 抗体组进行了基准测试,并与其中介绍的方法进行了比较。它在所有比较方法的前三分之一中执行。在长 CDRH3 的情况下,它最近与通用版本 RosettaAntibody 和 RosettaCM 进行了比较。令人惊讶的是,对于这些非常困难的案例,RosettaCM 比其他两种专用方法略好。
13 Biovia Discovery Studio/Antibody Modelling
BioVia Discovery Studio 在后端使用 Modeller 从用户提供的序列中对蛋白质结构进行建模。DS 基于同源性建模提出了不同的策略,但使用专用工具对抗体进行建模,称为生物治疗和抗体建模。它们在 AMI-I 和 -II 中进行了积极测试并得到了验证。
14 MOE/抗体管线
MOE 在两个数据库中搜索模板:内部数据库和 PDB 数据库。在自建数据库搜索过程中,如果与目标的序列相似度超过50%,模板将被保留。从内部构建的数据库和 PDB 数据库中,将分别保留前 10 名和前 5 名。然后通过嫁接框架和 CDR 来构建模板。建立多个模型,然后执行能量最小化。从这组模型中,建立共识模型,并提出不同的优化最终模型。
使用最终模型,进一步探索 CDR3 构象,最后进行聚类。一个独特的 CDR3 构象最终被移植到 V H H 模型中。再次进行了积极的评估。结合能最低的前三个模型将是该程序的最终输出。
15 薛定谔 BioLuminate 和抗体管道
BioLuminate是通用同源建模的管道,但包括抗体的特定设计。
建模协议是:
a)在精选的抗体数据库中检测框架,提供结构模板(使用序列身份选择)。
b)接下来,在扫描另一个仅包含 CDR 的自定义数据库和基于结构聚类、序列相似性和茎残基几何匹配的选择后,嫁接一组 CDR。
c)在最后一步,内部 Prime 软件重新包装侧链并最小化抗体模型。
d)使用 ab initio 方法构建 CDR3 循环。
Schrödinger 确实提供了有关 BioLuminate 的抗体建模工具和教程的详细信息(https://www.schrodinger.com/training/building-homology-models-multiple-sequence-viewereditor214,https://www.schrodinger.com/training/antibody-visualization-and-modeling-bioluminate-workshop-tutorial214,最后访问日期:2022 年 2 月 15 日)。
16 Macromoltek 的 SmrtMolAntibody
位于美国德克萨斯州奥斯汀的 Macromoltek 公司创建了 SmrtMolAntibody。该算法的第一步是搜索抗体数据库,以使用 BLAST 方法检测 VH 和 VL 查询序列的模板。然后,在loop数据库上进行类似的搜索,为所有六个 CDR 找到更合适的模板。基于确定的模板,组装框架和移植 CDR 允许构建初始抗体模型。对 CDR(H)3 loop进行了特定的建模步骤。保留了具有最佳(能量)CDRH3 环构象的前 50 个模型。对于这 50 种型号中的每一种,侧链都经过重新包装,并且它们的扭力被最小化。上述最终步骤基于能量评分进行多次评估,其中修改和软化的 Lennard-Jones 与侧链旋转异构体频率相关。
17 I-TASSER和C-I-TASSER
在多轮 CASP 中,通过 David Baker 的 Rosetta,I-TASSER是提出相关结构模型的最佳可用方法。I-TASSER 基于local Meta-Threading服务器 (LOMETS) 。LOMETS 利用基于深度学习的threading方法和基于配置文件的程序来识别最佳模板并注释基于结构的蛋白质功能。一旦确定了模板,I-TASSER 就会使用基于蒙特卡罗的模拟来生成和改进蛋白质结构模型。最新的发展包括 I-TASSER 方法中基于深度学习的接触预测,现在被命名为 CI-TASSER。
18 QUARK and C-QUARK
QUARK 是一种参考从头算结构预测策略。它基于由 11 个能量项组成的力场,经典地表示三个分辨率级别(原子、残差和拓扑)。
该策略可分为三个阶段:
根据输入序列,预测二级结构、扭转角和溶剂可及性以及生成的片段。然后,使用半还原蛋白质模型进行多副本交换蒙特卡罗 (REMC) 模拟,以构建多个替代构象。最后一步使用 SPICKER 进行聚类并对全原子结构进行细化。该软件的最新版本名为 C-QUARK。与 QUARK 相比,在开始时添加了两个步骤:(1)DeepMSA 生成的多序列比对和(2)基于深度学习方法的接触图预测。即使 QUARK 和 C-QUARK 在 CASP 比赛中表现良好。
19 AlphaFold2
AlphaFold 2 是 2020 年和 2021 年的热门话题,引发了蛋白质结构模型构建的一场革命。然而,AlphaFold 2 并不总是给出 100% 正确/有意义的预测,即,一些球状蛋白仍未正确建模,并且跨膜蛋白并不接近接近天然的结构,因为它们是难作用靶点(hard targets)。抗体没有经过专门测试[补充最近文献IgFold进行了测试,AlphaFold2依然是最好的方法,见前期公众号连接],但不同的例子表明,正如 Rosetta所见,经典的比较建模仍然表现更好。
20 RoseTTAFold and DeepAb
基于 AlphaFold 2 的特性和成功,Baker 的团队广泛研究了不同的神经网络架构,并提出了一种新的基于深度学习的方法,称为 RoseTTAfold。它基于三轨(1D、2D 和 3D)神经网络,可以同时处理多个序列比对 (MSA)、残基间接触和预测结构的细化。DeepAb可以认为是 RosettaAntibody 与深度学习 RoseTTAfold 的自然进化,该算法可以分为两个主要步骤。第一部分有一个深度残差卷积网络,它由 1D ResNet、一个 Bi-Long 短期记忆 (Bi-LSTM) 曲目编码器和 2D ResNet 组成。
21 NanoNet
NanoNet 于 2021 年 8 月向科学界提出。这确实是第一个优化的 V H H 方法。这种深度学习方法是使用经典抗体和 V H H训练的,因为需要大量数据来训练神经网络并获得相关结果。它的架构是由一个卷积神经网络(CNN)和两个一维残差神经网络(ResNet)组成的。该程序可在线获取(https://github.com/dina-lab3D/NanoNet,最后访问日期:2022 年 2 月 13 日)。
NanoNet 的评估是针对 AlphaFold 2 进行的,2021 年将 16 V H H 存储在 PDB 中,即没有参加 AlphaFold 2 训练。NanoNet 表现更好,平均 RMSD 为 2.69 (±1.49) Å,而 AlphaFold 2 为 3.23 (±2.49) Å(CDR3 为 1.57 (±0.41) Å vs. 2.04 (±2.09) Å,重复)。使用 Rosetta Antibody 建模套件时,使用 37 V H H获得了类似的结果。NanoNet 表现更好,平均 RMSD 为 1.68 (±0.57) Å vs. Rosetta Antibody 的 2.71 (±1.13) Å(CDR3 分别为 2.99 (±1.48) Å vs. 5.73 (±2.33) Å)。
抗体结构预测相关文章
Johns Hopkins | 可与AlphaFold2准确率媲美的更快的抗体结构预测工具IgFold
ICLR 2022|迭代精调的GNN用于抗体-序列结构协同设计
回台回复: antibody 获取文献