蛋白质-蛋白质相互作用(PPI)对正常的细胞功能至关重要,并且与许多疾病途径有关。一系列蛋白质功能由蛋白质相互作用通过翻译后修饰(PTM)介导和调节。
然而,在 IntAct 等生物知识数据库中,只有 4% 的 PPI 使用 PTM 进行注释,主要通过人工管理进行,既不省时也不划算。研究人员的目标是通过使用深度学习的远程监督训练数据来帮助人类管理,从文献中提取 PPI 及其成对 PTM 来促进注释。
墨尔本大学的研究人员
使用 IntAct PPI 数据库创建一个远程监督数据集,该数据集标注了相互作用的蛋白质对、它们相应的 PTM 类型以及来自 PubMed 数据库的相关摘要。
他们
训练了一组 BioBERT 模型(称为 PPI-BioBERT-x10)以提高置信度校准;扩展了具有置信度变化的整体平均置信度方法的使用,以抵消类不平衡的影响,以提取高置信度预测。
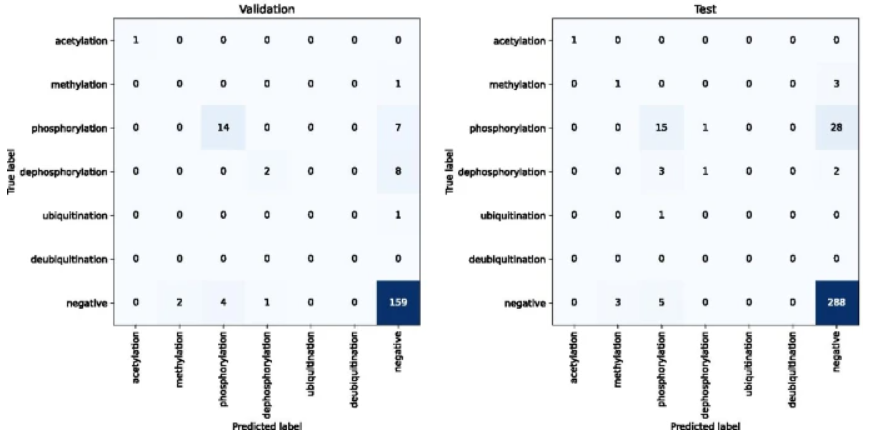
在测试集上评估的 PPI-BioBERT-x10 模型产生了适中的 F1-micro 41.3(P = 58.1,R = 32.1)。然而,通过结合高置信度和低变化来识别高质量的预测,调整预测的精度,研究人员以 100% 的精度保留了 19% 的测试预测。
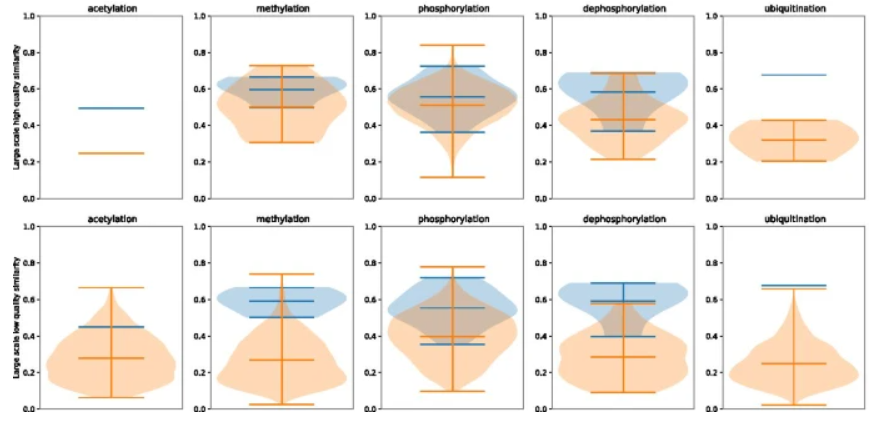
他们在 1800 万份 PubMed 摘要上评估了 PPI-BioBERT-x10,提取了 160 万个PTM-PPI 预测,并过滤了大约 5700 个高置信度预测。在 5700 个中,对一个小的随机抽样子集的人工评估表明,尽管进行了置信度校准,但精度仍下降到 33.7%,并且即使进行了置信度校准,也凸显了超出测试集的通用性挑战。
研究人员通过只包括与多篇论文相关的预测来规避这个问题,将精度提高到 58.8%。在这项工作中,他们强调了基于深度学习的文本挖掘在实践中的好处和挑战,以及需要更加强调置信度校准以促进人工管理工作。
该研究以「Large-scale protein-protein post-translational modification extraction with distant supervision and confidence calibrated BioBERT」为题,于 2022 年 1 月 4 日发布在《BMC Bioinformatics》。
关键的生物过程,例如信号级联和新陈代谢,受到蛋白质-蛋白质相互作用(PPI)的调节,这些相互作用通过修饰其他蛋白质以通过翻译后修饰(PTM)调节它们的稳定性或活性。PPI 在大型在线存储库(例如 IntAct 和 HPRD)中进行管理。
然而,大多数 PPI 没有用函数注释,例如,IntAct 数据库有超过 100,000 个人类 PPI,但其中不到 4000 个用 PTM 注释,如磷酸化、乙酰化或甲基化。了解相互作用的蛋白质对之间 PTM 的性质对于研究人员确定网络扰动和下游生物学后果的影响至关重要。
生物数据库中的 PPI 和 PTM 通常是手动管理的,这需要训练有素的管理人员,同时也很耗时。科学家还强调了维护手动管理数据库、确保它们是最新的以及手动管理的经济方面的其他挑战。因此,采用自动策展方法对于这项工作的可持续性至关重要。
研究人员通过文本挖掘 PubMed 摘要来提取 PTM,提取蛋白质对及其相应的 PTM。给定输入期刊摘要,输出是 <Protein1, PTM function, Protein2> 形式的三元组,其中 Protein1 和 Protein2 是蛋白质的 Uniprot 标识符。由于他们的训练数据源不包含蛋白质之间关系的方向,所以不考虑 Protein1 和 Protein2 之间的关系方向,即 <Protein1, PTM function, Protein2> 等价于 <Protein2, PTM function, Protein1>。
研究人员还旨在帮助 PTM-PPI 的人工管理,因此他们通过将机器学习模型应用于 1800 万个 PubMed 摘要以提取 PTM-PPI 三元组来评估机器学习模型的泛化程度。在这里,研究人员使用置信度校准作为一种机制来理解通用性,以了解预测何时可以提取高质量的预测。该团队相信他们是第一个研究使用具有深度学习和远程监督的 NLP 进行大规模 PTM-PPI 提取的实际适用性和挑战的团队。
研究人员专注于提取 PTM,包括磷酸化、去磷酸化、甲基化、泛素化、去泛素化和乙酰化(这些 PTM 是根据训练数据的可用性选择的)。他们使用远程监督方法创建一个训练数据集,使用 IntAct 作为源知识库从 PubMed 摘要中提取 PTM-PPI 三元组。
他们训练了一组 BioBERT 模型来改进神经置信度校准。然后,将经过训练的模型应用于 1800 万份 PubMed 摘要,以提取 PPI 对及其相应的 PTM 函数;并尝试使用神经置信度校准技术来确保高质量的预测,以增强和促进人类管理工作。
用于评估文本挖掘方法的 PPI 提取数据集(例如 AIMed 和 BioInfer)在十多年来(自 2007 年以来)一直保持不变,并且专注于提取蛋白质相互作用,而不是它们之间 PTM 相互作用的性质。这些数据集还被用于评估最新的机器学习方法,包括蛋白质对提取中的深度学习。
然而,在使用 AIMed 和 BioInfer 数据集的基准测试方法的有限背景下,最新的深度学习趋势似乎在 PPI 管理中并不广泛流行。使用文本挖掘和基于规则的方法的自动 PPI 管理尝试似乎更普遍。
PPI-BioBERT-x10 上测试和验证集的混淆矩阵。
STRING v11 是最受欢迎的 PPI 数据库之一,它使用文本挖掘作为策展方法。自 STRING v9.1 以来,他们的文本挖掘管道基本保持不变。STRING v9.1 使用基于加权 PPI 共现规则的方法,其中权重取决于蛋白质对是否一起出现在同一文档、同一段落或同一句子中。即使训练数据有限,基于规则的方法也可能非常有效,具体取决于任务。
将 STRING v11 数据库中的交互单元定义为「功能关联,即两种蛋白质之间的联系,它们共同促成特定的生物学功能」。这个定义允许基于共现规则的方法非常有效,即如果一个蛋白质对经常在文本中同时出现,那么这对很可能是相关的。
iPTMnet 从各种手动管理的数据库(例如 HPRD 和 PhosphoSitePlus 以及文本挖掘资源)中整合有关 PPI 和 PTM 的信息。对于文本挖掘,iPTMnet 使用 RLIMS-P 和 eFIP 来自动管理酶-底物-位点关系。这些工具使用基于规则的方法,使用文本模式来提取 PTM 中涉及的蛋白质。
2019 年 11 月的 iPTMnet 统计数据表明,使用 RLIMS-P 策划的酶-底物对总数少于 1,000 对。这个适度的数字突出了使用文本模式的主要挑战:虽然它们可以以相当高的精度提取关系,但它们对于如何在文本中描述 PPI 关系的变化并不稳健。因此,研究人员探索了基于机器学习的方法,这些方法能够提取更多的关系。
使用深度学习自动提取 PPI 可能是有益的,因为它有可能从各种文本中提取 PPI,其中 PPI 关系的描述方式无法通过手动制作的基于规则的系统轻松捕获。
然而,深度学习需要大量的训练数据。确保预测质量的模型的通用性是其广泛采用从文本中自动提取 PPI 关系的关键。大规模提高预测质量需要专注于减少误报,以最大限度地减少对现有知识库条目的破坏,因此,减少低质量预测的置信度校准方法成为大规模文本挖掘的关键步骤。
置信度校准是预测代表真实正确性的概率估计的问题,在这里,研究人员使用置信度校准来了解预测何时可能正确,并将其用作改进泛化的机制。通用性的方面在很大程度上仅限于对测试集的评估,而使用测试集性能作为现实世界性能的代理的局限性在以前的研究中受到了挑战。
创建具有细粒度注释的黄金标准训练数据,是一项手动的劳动密集型任务,并且是将机器学习应用于新领域或任务的限制因素。能够利用一个或多个现有数据源是在新领域或新任务中使用机器学习的关键。远程监督利用现有的知识库,例如 IntAct,而不是注释新数据集。
然而,使用远程监督数据集有两个主要限制:(a)噪声标签需要降噪技术来提高标签质量(b)它们需要生成负样本,因为数据库通常只包含关系的正样本。
BiLSTM 和 BioBERT 等深度学习架构以前已用于使用自然语言处理(NLP)和 AIMed 数据集对蛋白质关系提取方法进行基准测试。
然而,这些工作并没有衡量这些模型校准置信度分数的能力。研究人员选择了最先进的深度学习方法 BioBERT,训练一个集成来增强置信度校准,并使用置信度变化来抵消置信度校准期间类不平衡的影响。
论文链接:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04504-x
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。