计算生物学揭秘奥密克戎强感染性原因

(本文阅读时间:10分钟)
近年来,微软亚洲研究院在新冠病毒方面进行了一系列研究。
2021 年,微软亚洲研究院与清华大学在《Advanced Theory and Simulations》上共同发表了封面文章《通过分子动力学模拟探索 SARS-CoV-2 刺突蛋白 NTD 的调控功能》(Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein Through Molecular Dynamics Simulation)[1]。
这是微软亚洲研究院和清华大学生命科学学院龚海鹏教授团队共同开展的一项计算生物学研究,主要通过分子动力学模拟、研究新冠病毒侵染人类机理中 N 端结构域 NTD(N-Terminal Domain)的作用。
据了解,在新冠病毒表面有一层 S 蛋白,只有在被人体的受体蛋白 ACE 2 识别以后,病毒才能进入人体。两者结合能力越强,就越容易造成感染。
S 蛋白的形态有点像字母“Y”,向下的一竖即 S2 区域嵌入在病毒表面,起到固定的作用。伸出来的两个“枝丫”分别叫作 NTD 和 RBD(受体结合域,Receptor Binding Domain)。
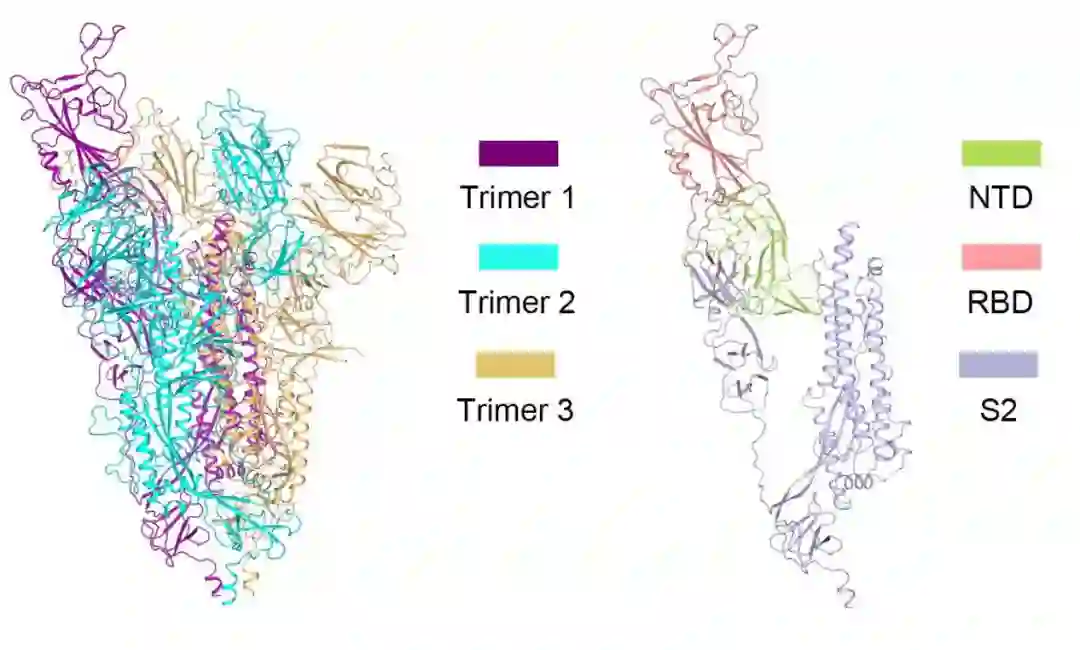
图1:新冠病毒 S 蛋白结构(左图为 S 蛋白三聚体结构,由三条链组成;右图为 S 蛋白单体结构,由 NTD、RBD 以及 S2 组成)
在该研究之前,人们已经通过实验知道了 RBD 可以直接起到一个识别人体受体蛋白的作用,而 NTD 的具体作用还不十分清晰。
微软亚洲研究院和龚海鹏教授实验室合作搭建了一个有百万个原子的稳定模拟体系,在进行了数十亿步(1 步是 1 飞秒,即一千万亿分之一秒)的分子动力学模拟之后,提出了一个“楔形”模型假说,简单来说,可以把 NTD 看作是安插在了 RBD 和“Y”字底部的一个“楔子”。
研究发现,RBD 有 down 和 up 两种状态,只有 up 状态才有可能与人的受体蛋白做识别。该项研究发现,RBD 倾向于从 up 的状态转变为 down 的状态,而当 NTD 像楔子一样塞入 RBD 下端时,阻止了 RBD 的这一倾向,即 NTD 使得 RBD 保持 up 状态,进一步让其拥有了感染的能力。
可以说,NTD 在病毒的侵染过程中起到了一个调控作用,调节 RBD 的形态,从而改变了病毒感染人体的倾向。
图2:NTD 在 SARS-CoV-2 的 S 蛋白构象变化中发挥调控功能示意图
由此可以预见,NTD 调控 RBD 的界面可能成为潜在的药物靶点。若能设计一个药物分子可以把 NTD 向外拉出,也就是把这个“楔子”拔出来,那 RBD 就倾向于 down 的状态,变成没有感染能力的构象,病毒感染人的能力就会减小很多。
“我们这项工作在世界范围内首次提出了 NTD 在病毒侵染过程中的调控模型。此外,我们根据这个潜在的药物靶点,设计了药物的虚拟筛选算法,并对新冠病毒的一些潜在性药物做了一定筛选,为新冠病毒药物研发提供了一定的参考价值。”微软亚洲研究院主管研究员王童说道。
近期,微软亚洲研究院和清华大学生命科学学院王新泉教授和医学院张林琦教授课题组开展合作,在奥密克戎变异株强传染性的机理解释方面有了新的突破,其成果已被生物学领域的顶级期刊《Cell Research》接收。
据了解,在此次合作中,王新泉教授课题组率先解析了奥密克戎高分辨率的晶体结构,为奥密克戎感染机理研究奠定了基础。
相比于静态的晶体结构,王童团队利用分子动力学模拟从“动态视角”模拟和分析奥密克戎结构变异及其侵染机理。
研究中,微软亚洲研究院的研究员们首先构建了两个模拟体系,分别以原始的新冠病毒结构和奥密克戎的结构作为起始结构,对这两个结构平行地进行长时间、数亿步的分子动力学模拟,模拟出的是原子级别的运动过程,从而观察病毒在人体中的真实变化。
(来源:Pixabay)
“对分子运动进行模拟的方法可以分成两种,经典的分子动力学模拟和基于第一性的量子模拟,”王童说,“对于研究新冠病毒中蛋白大分子、大尺度的构象变化运动,经典动力学模拟方法是一种更适合的手段。”
据王童介绍,经典模拟适用于大体系,像蛋白质成千上万的原子,比如此次模拟体系就包含上百万的原子。从时间上来说,经典模拟做的都是数亿到数十亿步的模拟。相比之下,量子模拟相对准确,但计算过程的耗时使其只能适用于非常小的体系,比如对只有十余个原子的体系计算性质或进行短暂的模拟。
构建了模拟体系之后,微软亚洲研究院利用自研算法,分析了原始新冠病毒和奥密克戎感染人体的能力,以及它们结构上非常细微的差异。
值得注意的是,研究过程中产生的数据量极其庞大,整个模拟下来,至少有数亿个结构,产生了数亿到数十亿帧的结构变化。
为此,研究人员创造性地提出一种全新的马尔科夫模型算法对数据进行分析。其对分子动力学模拟的数据分析有着更强的表征能力,可以更加真实地模拟出蛋白的动态变化过程。
在对海量的数据进行筛选和分析后,研究人员将数十亿的结构聚合成几类代表性的结构,再去分析它们之间的区别和如何相互转换,以及与人受体蛋白的结合能力,从而更好地理解奥密克戎感染的分子机理。
这些聚合出来的代表性构象被称为“亚状态”,研究人员对其做了定性的结合自由能计算,以此在一定程度上反映病毒的感染能力。
具体来说,在研究中,聚类以后的奥密克戎和原始病毒都有三个亚状态。由于奥密克戎本身只是原始病毒的变体,所以它们各自占比最高的一个亚态,无论是从结构上,还是和人的受体蛋白结合的能力上来说都是非常类似的。
由此可见,奥密克戎结构的关键不同之处在其他两个亚状态上。相比原始病毒的两个亚状态,奥密克戎的两个亚状态和人的受体蛋白ACE2有更多的相互作用,结合能力更强(结合自由能更低),因此可能导致了更强的感染能力。
同时研究还发现,奥密克戎的三个亚状态之间相互转换非常快,很容易从类似于原始病毒的主亚状态,转换成结合能力更强的另外两个亚状态。这也从动态视角解释了为什么奥密克戎感染性这么强的一个原因。
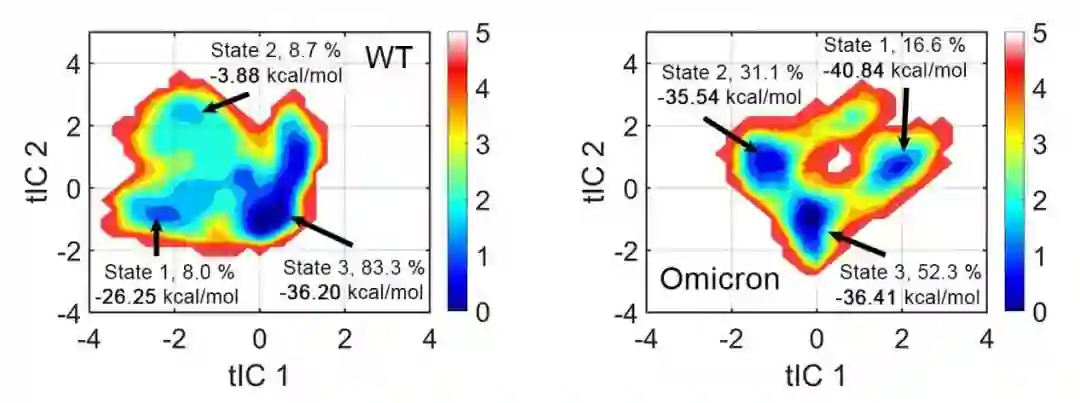
图3:分子动力学模拟揭示奥密克戎的状态变化及感染机理(左图为新冠病毒 S 蛋白,右图为奥密克戎变异株的 S 蛋白,其中两个体系的 State3 基本一致且为主要构象,奥密克戎的另外两个亚态结合自由能明显高于新冠病毒的两个亚态)
在最新有关奥密克戎的研究工作中,王童表示,“这项研究在世界上首次使用‘干湿结合’的方式和动态视角,从结构生物学和计算生物学两个角度同时出发,提出了奥密克戎感染性强的分子机理。”
上面提到的“干湿结合”,也就是“干实验”(计算实验)和“湿实验”(传统科学实验)的结合。
据了解,“干实验”可以对“湿实验”现有的一些结论做验证和补充,还能做一些“湿实验”做不了的或者是需要花很多时间、人力去做的研究。
比如,在奥密克戎的研究中,若在“湿实验”实验室里一个个去分析突变位点结构的变化,可能需要数月时间,在疫情如此严峻的情况下,显然不利于疫情防控工作。而使用计算的手段来模拟这种突变造成的影响,可能只需要一周到两周的时间。
另外,“湿实验”在前期很多时候都是探索性质的,甚至在研究方向上是非常迷茫的。这时“干实验”可以在一定程度上引导“湿实验”的实验设计,帮助其找到一个更容易成功的方法。
值得注意的是,两者并不是互相取代,而是同等重要、相互指导、相辅相成的。“我们在和研究伙伴合作的过程中就有这样的感觉,我们认为‘干湿结合’会对整个科学研究起到一个螺旋式上升、波浪式前进的推动作用。”王童说道。

微软亚洲研究院主管研究员王童
他还补充说,在新冠病毒的研究方面,与清华大学多位老师的合作是一个非常好的起点。微软亚洲研究院非常希望能和海内外高校、科研院所的专家学者们有更多的深入合作和跨领域交流。
据了解,微软亚洲研究院后续还会就新冠病毒为何在整个冠状病毒进化过程中感染性如此强的原因方面开展相关研究。
最后,关于计算生物学等跨学科领域对人才的要求和发展趋势,王童也发表了自己的看法。
他认为跨学科发展主要是围绕一个关键词,就是“交叉”。交叉不是简单的“加法”,而是“乘法”。两个学科之间要发生一定的化学反应和深度的融合。
拿计算生物学来说,其本质上就是计算机科学与生物学的交叉,需要结合两个领域的知识,并真正消化理解,才能在计算生物学领域产生真正的学术影响力,解决真正的科学问题。
而这也对人才的培养或者说对人才的需求,提出了一个更高的要求。如果研究人员只掌握某一方面的知识,对另一学科的认识不够深入,只停留在简单的工具使用层面,那么研究工作就只能浮于表面,也很容易造成计算参数设置有误等问题,导致最终计算结果与实验结果完全不一样的情况。
未来,各学科之间的交叉融合一定会越来越多,在具体的科研工作中,不同背景的人才在合作交流过程中,需要更多倾听对方领域的知识和诉求,只有建立在互信、开放心态的基础上,才能推动跨学科间的研究持续向前发展。
参考:
1. Y. Li,T. Wang,J. Zhang,B. Shao,H. Gong,Y. Wang,X. He,S. Liu,T. Liu.Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein through Molecular Dynamics Simulation.Advanced Theory and Simulations 4(2021).
https://doi.org/10.1002/adts.202100152
你也许还想看:









