武汉肺炎疫情新型冠状病毒最新研究: 2019-nCoV资源库发布与基因组生物信息学分析
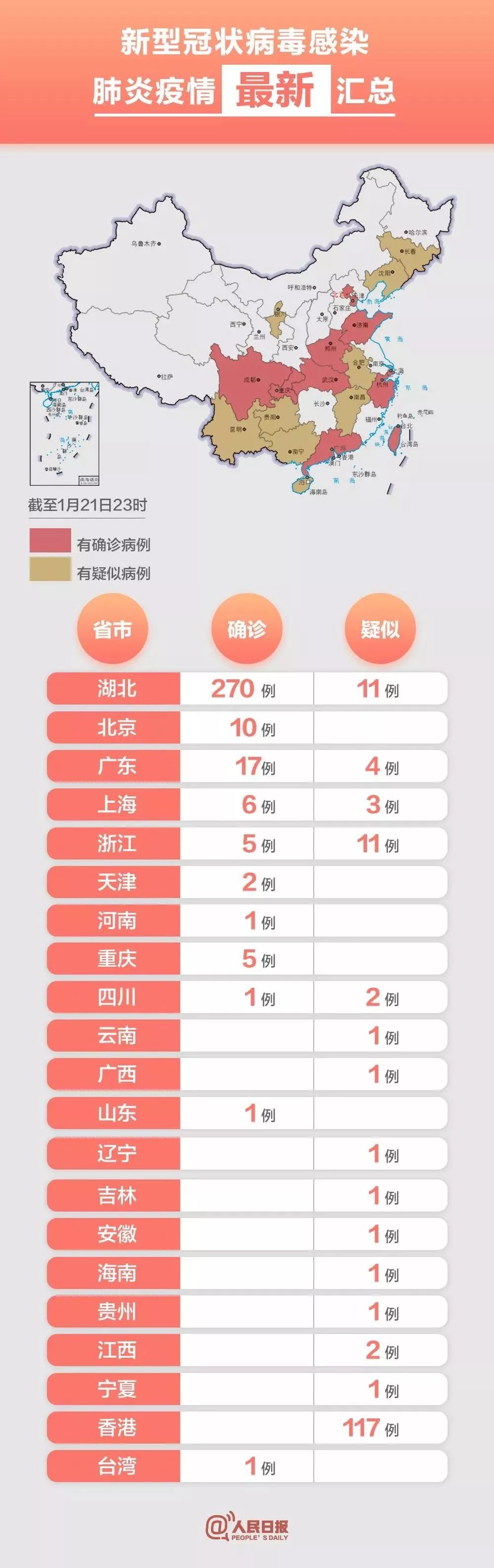
武汉新型冠状病毒(2019-nCoV)引起的肺炎疫情蔓延牵动春节你我。据国家卫健委统计,截至1月22日24时,收到国内25个省(区、市)累计报告新型冠状病毒感染的肺炎确诊病例571例,其中重症95例,死亡17例(均来自湖北省)。13个省(区、市)累计报告疑似病例393例。
对于我们普通人做好:一 外出戴好口罩,二没事宅在家里,三时刻勤洗手 四不造谣不传谣,此外我们要相信科学,相信医生,相信知识,众志成城,共克时艰。我们来看下最新一些关于新型冠状病毒研究进展:
1. 国家基因组科学数据中心发布2019新型冠状病毒资源库
2020年1月22日,国家基因组科学数据中心正式发布2019新型冠状病毒资源库。该库整合了世界卫生组织(WHO)、中国疾病预防控制中心(CDC)、美国国家生物技术信息中心(NCBI)、全球流感序列数据库(GISAID)等机构公开发布的冠状病毒基因组序列数据、元信息、学术文献、新闻动态、科普文章。同时,对不同冠状病毒株的基因组序列做了变异分析与展示。
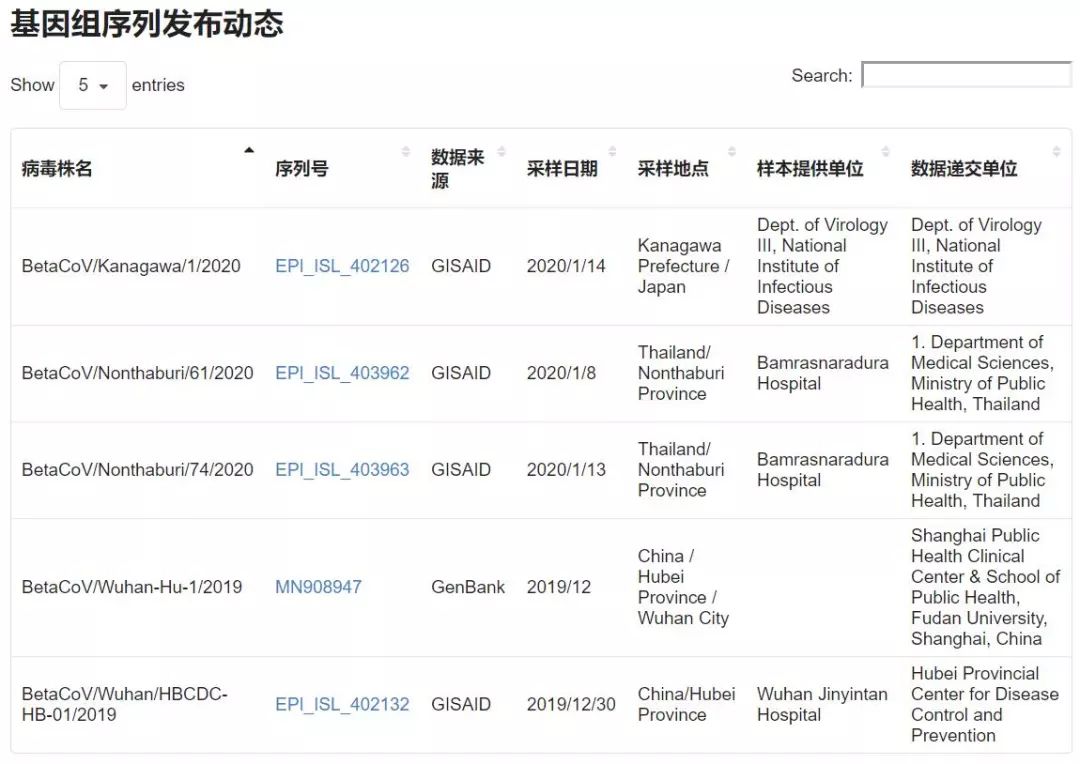
2019新型冠状病毒基因组序列发布动态
2019新型冠状病毒资源库收录了来源于NCBI的GenBank数据库和GISAID数据库发布的2019新型冠状病毒(2019-nCoV)病毒株的株名、采样日期、采样地点、样本提供单位、数据递交单位等元信息。通过该资源库还可访问到国家基因组科学数据中心基因组数据库GWH从公共数据库收录的冠状病毒科基因组和蛋白序列,用户可基于Accession号、种名、属名、采样日期、采样地点、宿主、分离源、发布日期等元信息筛选感兴趣的冠状病毒株,个性化选取序列进行下载以开展相关的科学研究。
2019新型冠状病毒资源库将持续更新元信息与基因组序列数据,实时监控NCBI的PubMed数据库中发表的2019新型冠状病毒和其他冠状病毒的学术文献、中新网与新华网发布的新闻,同步更新世界卫生组织与中国疾病预防控制中心发布的科普介绍,为用户开展学术研究、掌握科研进展、了解新闻动态与科学知识提供资源与窗口。
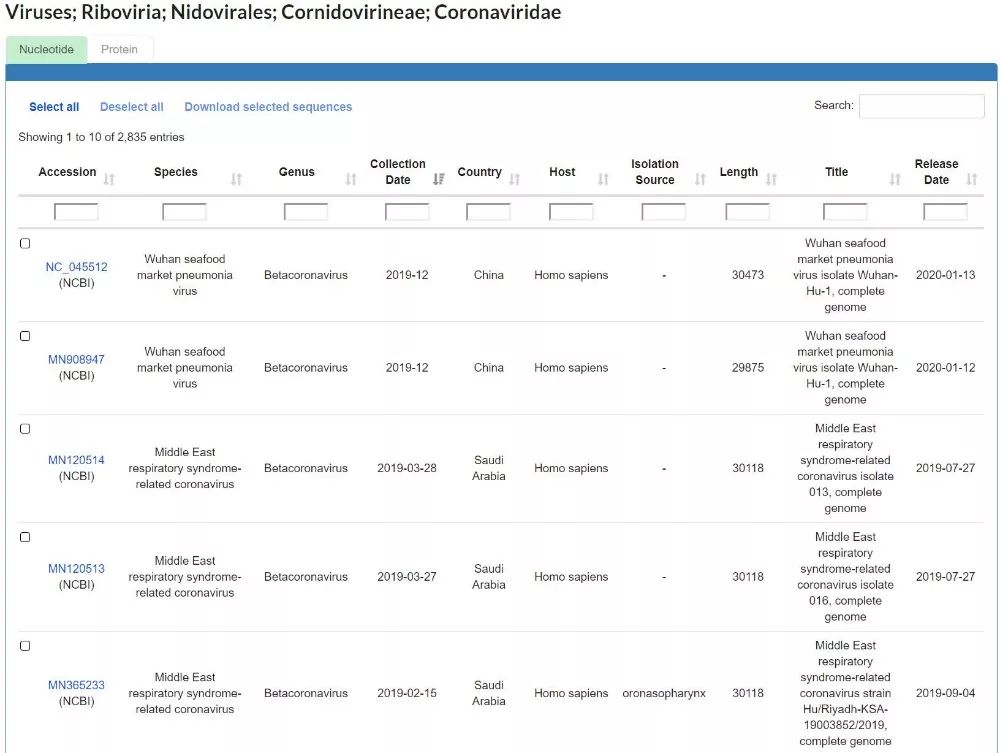
GWH数据库收录的冠状病毒科基因组序列信息
2019新型冠状病毒资源库基于不同参考基因组序列开展2019-nCoV病毒株基因组变异分析,并对结果进行了统计与可视化展示。通过对全基因组序列相似性比较和变异位点分析,获取2019-nCoV病毒株之间、2019-nCoV病毒株与SARS冠状病毒以及与类SARS冠状病毒蝙蝠株之间的变异程度、变异区域、变异碱基的详细信息。经数据分析,2019-nCoV与2003年爆发的SARS病毒基因组序列相似度为80%,与2017年2月从国内的蝙蝠中采集到的Bat SARS-like coronavirus isolate bat-SL-CoVZC45基因组序列相似性最高,相似度为88%。对2019-nCoV病毒株的基因组变异分析为追溯病毒来源、追踪病毒株变异路径、防控新型冠状病毒引发的疫情、治疗病毒性肺炎提供重要的数据基础与决策支持。
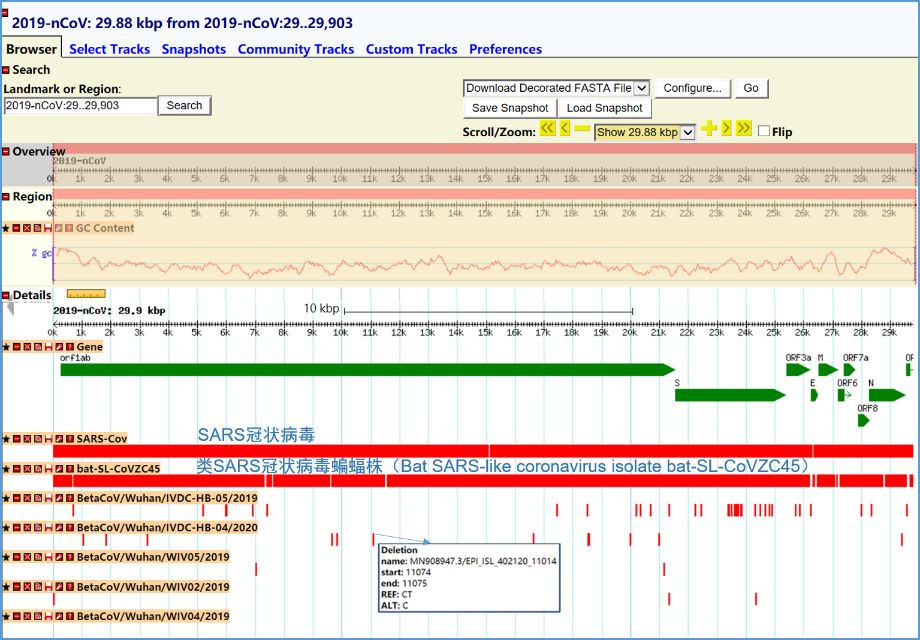
2019新型冠状病毒基因组变异分析:基因组变异分析的参考基因组为GenBank发布的2019新型冠状病毒株基因组序列MN908947.3,SARS-Cov为SARS 冠状病毒,bat-SL-CoVZC45为类SARS冠状病毒蝙蝠株,其余基因组序列来源于GISAID发布的2019新型冠状病毒株。红色竖线表示变异位点。
参考链接:
http://www.big.cas.cn/xwzx/kyjz/202001/t20200122_5494163.html
2. 北京大学等多机构联合攻关,首次发现蛇是最有可能的携带2019-nCoV病毒的野生动物
2020年1月22日,北京大学,广西中医药大学,宁波大学及武汉生物工程学院学者联合攻关,在Journal of Medical Virology 在线发表题为“Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross‐species transmission from snake to human”的研究论文,为了确定可能的病毒库,该研究基于新发现的冠状病毒2019-nCoV的现有序列,结合不同动物物种之间的相对同义密码子使用偏倚(RSCU)情况进行了全面的序列分析和比较。分析中获得的结果表明,2019-nCoV似乎是蝙蝠冠状病毒与起源未知的冠状病毒之间的重组病毒。重组发生在病毒spike糖蛋白内,该蛋白识别细胞表面受体。此外,该研究结果表明,与其他动物相比,基于蛇的RSCU偏差类似,蛇是最有可能的携带2019-nCoV病毒的野生动物。

参考链接:
https://onlinelibrary.wiley.com/doi/10.1002/jmv.25682
3. Nature: 病毒传播不能忽视流体力学
针对目前中国武汉发生的病例,这种新型冠状病毒的直径约为0.1um,但因为其需要附着在唾液等飞沫上,所以载体的空气动力学直径要大于2.5um。
4. 中科院最新论文「武汉新型冠状病毒」的进化来源和传染人的分子作用机制和传染性风险
2020年1月21日,中国科学院上海巴斯德研究所郝沛研究员、军事医学研究院国家应急防控药物工程技术研究中心钟武研究员和中科院分子植物卓越中心合成生物学重点实验室李轩研究员合作,在SCIENCE CHINA Life Sciences(《中国科学:生命科学》英文版),在线发表了题为“Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission”的论文,为武汉新型冠状病毒的进化来源和传染人的机制给出了学术解释。
该论文分析阐述了引起近期武汉地区肺炎疫情爆发的新型冠状病毒的进化来源,及与导致2002年广东“非典”疫情的SARS冠状病毒、“中东呼吸综合征”MERS冠状病毒的遗传进化关系,并通过对武汉的新型冠状病毒spike-蛋白的结构模拟计算,揭示了武汉新型冠状病毒spike-与人ACE2蛋白作用并介导传染人的分子作用通路。该成果评估了武汉新型冠状病毒的潜在人间传染力,为尽快确认传染源和传播途径、制定高效的防控策略提供了科学理论依据。

5. 南开大学-武汉2019冠状病毒基因组的生物信息学分析
武汉2019冠状病毒基因组的生物信息学分析
陈嘉源施劲松丘栋安刘畅李鑫赵强阮吉寿高山
南开大学生命科学学院东部战区总医院英国诺丁汉特伦特大学生物科学系南开大学医学院南开大学数学科学学院
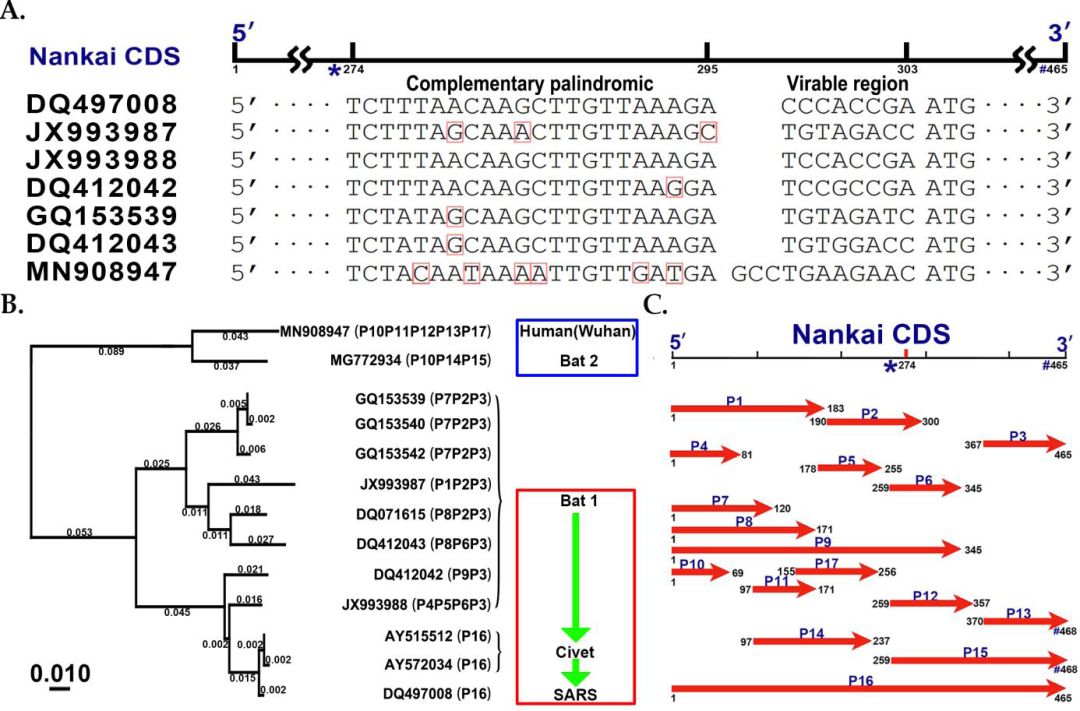
2019年12月,中国武汉报道了冠状病毒引起的肺炎,其临床症状与2003年爆发的严重急性呼吸综合征(Severe Acute Respiratory Syndrome, SARS)不同,因此推断该病毒可能是冠状病毒的一个新变种。不同于简单使用全基因组序列的其它研究,我们于2018年在国际上首次提出分子功能与进化分析相结合的研究思想,并应用于冠状病毒基因组的研究。在这一思想指导下,本研究使用beta冠状病毒基因组中的一个互补回文序列(命名为Nankai complemented palindrome)与其所在的编码区(命名为NankaiCDS)对新发布的武汉2019冠状病毒基因组(GenBank:MN908947)进行分析以期准确溯源,并对beta冠状病毒的跨物种传播和宿主适应性进行初步研究。溯源分析的结果支持武汉2019冠状病毒源自中华菊头蝠,但与SARS冠状病毒差异巨大,这一结果与两者临床症状差异一致。本研究的最重要发现是beta冠状病毒存在大量的可变翻译,从分子水平揭示了该病毒变异快、多样性高的特点,为beta冠状病毒的防控提供了依据。从beta冠状病毒可变翻译中获取的信息可应用于(但不限于)其快速检测、基因分型、疫苗开发以及药物设计。另外,我们推断beta冠状病毒可能通过可变翻译以适应不同宿主。本研究是基于大量基因组数据的实证分析,在国际上首次从分子水平尝试解释了beta冠状病毒变异快、宿主多且具有较强的宿主适应性的原因。
参考链接:
https://kns.cnki.net/KCMS/detail/23.1513.Q.20200120.0839.002.html
便捷查看下载,请关注专知公众号(点击上方蓝色专知关注)
后台回复“NCOV” 就可以获取《新型冠状病毒相关文献》相关文献下载链接索引