诺奖级成果开源!为什么说AlphaFold2足以改变全人类?

文 | 炼丹学徒
编 | 小轶
前天,AlphaFold2开源,相信大家被大大小小的公众号刷屏了。谷歌Deepmind团队此前使用基于Transformer的模型,在CASP14比赛上,刷新蛋白质三维结构预测的新高度,而详细论文,代码,久久没有开源,大家翘首以待到现在,终于等来了它。为什么大家都是魔改Transformer刷SOTA,别人万众瞩目Nature抢着发,被生化医药圈子里的科研人员认为是诺奖级别的工作,然而我们也是用Transformer天天刷SOTA,却发个会议都感觉无比艰难呢?
鉴于大家都是搞NLP的不懂生物,对于别人热火朝天的AlphaFold2讨论感觉自己格格不入,为了解决这个问题,卖萌屋从NLP研究员的角度,看一看这个被Transformer轰动一时的蛋白质三维结构预测为何如此有影响力,通过浅显但完整的知识介绍,带大家无痛入门生物大分子。因为炼丹学徒本人也是做NLP的,具体的生物学知识的细节可能存在错误,希望大家如果读到可以指出。希望抛砖引玉,通过阅读本文可以花费很短的阅读时间,建立起所需要的知识大体框架,如果有进一步了解的兴趣,再去进一步调研相应的部分。
本篇观点文章的结构组成为,蛋白质结构预测的任务介绍,对人类的意义,蛋白质结构预测的经典算法和近期NLP+蛋白质的相关工作介绍,AlphaFold2的突破之处和具体做法,以及总结和展望。
论文题目:
Highly accurate protein structure prediction with AlphaFold
开源链接:
https://github.com/deepmind/alphafold
论文链接:
https://www.nature.com/articles/s41586-021-03819-2_reference.pdf
另有论文算法附录[1]和博客[2]放在文末参考文献中
Arxiv访问慢的小伙伴也可以在 【夕小瑶的卖萌屋】订阅号后台回复关键词 【0718】 下载论文PDF~
蛋白质结构预测的任务介绍和所需要的基础知识
蛋白质结构
首先我们看一下蛋白质三维结构预测的任务定义。输入氨基酸序列,预测其三维结构坐标,此处,三维结构坐标指的是氨基酸组成的蛋白质中,每个原子的x, y, z坐标。我们是碳基生物,氨基酸的中间也是一个碳原子,碳原子有四个未配对的游离的点子,因此可以连出去四个单键,分别是羧基COOH,氨基NH2,一个氢原子H和自由基R,R的不同决定了这个氨基酸的不同,常见的可以组成生命的R只有20种,因此我们做NLP的可以把它当做一个21个词的词典给他embedding(其他不常见的丢进第21个词UNK)。氨基酸的COOH和NH2可以脱水缩合成肽键CONH把很多的氨基酸连成一个长链,称为肽链。肽链中的氨基酸由于连接时脱水缩合失去了一分子水H2O不是完整的氨基酸,因此称为残基,我们在读生物学论文里常见到的residue,指的就是它。
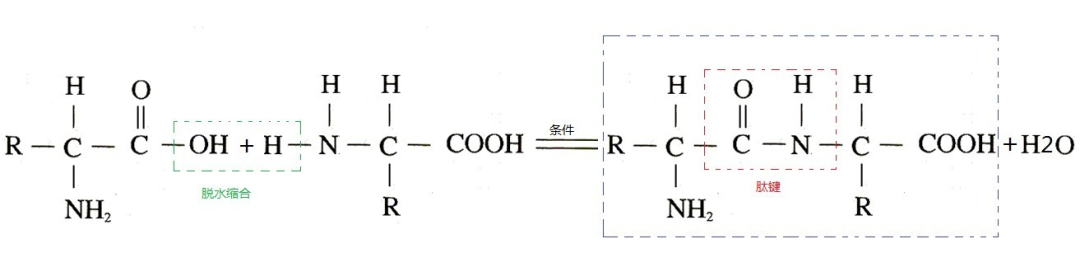
因为这个NH2-CH-COOH是固定结构(R是不固定的),而这个氨基酸的COOH要和下一个氨基酸的NH2结合成肽键连起来,因此这一条连续的肽键-碳-肽键上的原子是一定会存在的,这些碳、氧、氮的链称为骨架链(backbone),而那些不同的R被称为侧链(side chain)。氨基酸的顺序被称为氨基酸的一级结构。二级结构是折叠后规则的片段,周期性的结构构象,比如挨着的肽键会形成某一个角度的螺旋上升( 螺旋),一级结构上挺远的片段通过氢键折叠在一起( 折叠),或者松散的结构(coil),如果某个位置发生了90度以上的急转弯,则又记录为 转角。三级结构则是考虑上那些侧链里更复杂的作用关系后的完整三维坐标,比如二硫键把某些位置拉近了,coil结构到底松散成了什么具体形状,某些作用力又使得结构更复杂地变化了。两个以上的蛋白质可以通过分子对接可以拼成更复杂的结构,也称为四级结构,对接的过程被称为docking,是信号分子发生作用,蛋白质发挥功能,药物和蛋白质结合等生化反应里的重要模拟。
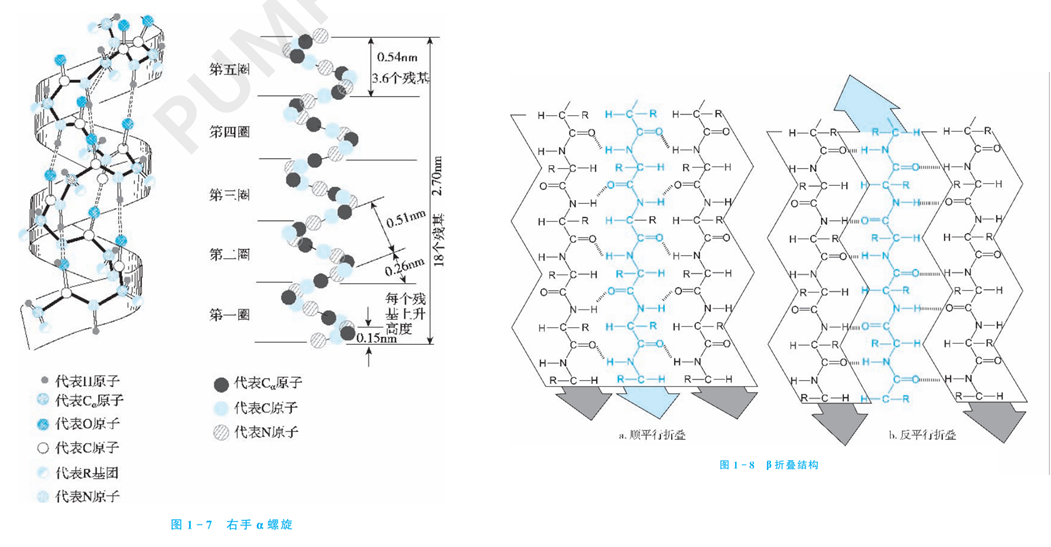
生物学上认为,低级的空间结构可以决定高级的空间结构,即仅仅输入氨基酸序列(一级结构),是可以预测出其空间结构的。对我们做NLP的来说,预测二级结构在模型上最简单,就是序列标注,对每个氨基酸分类在 螺旋区域,还是 折叠等等,就好比在BERT的最后一层做一个八分类。预测三维结构则复杂一些,需要预测到每个氨基酸甚至是氨基酸内部原子的坐标,其计算方法我们等下再谈。
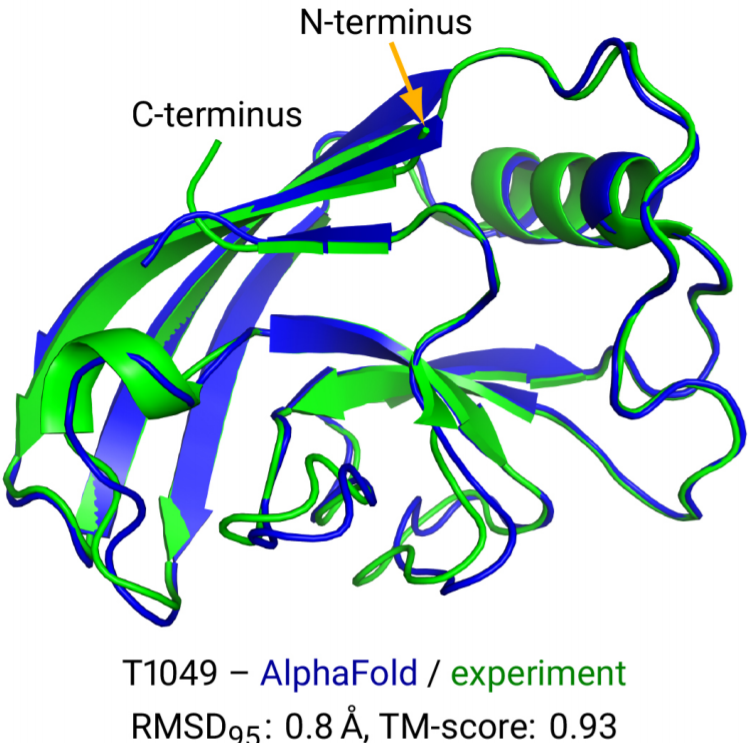
比如AlphaFold2论文里预测的这个蛋白质结构,螺旋的部分就是 螺旋比如右上部分,宽箭头部分是 折叠比如图片中间和左上部分,松散的线是coil部分如图片的右下部分。可以看到,二维结构可以一定程度上描述蛋白质的空间结构,但是如果具体的描述出来其形状,还是需要三维结构的坐标描述。这个坐标的原点和方位是任意的,即随意旋转之后表达的仍然是相同的蛋白质。
MSA

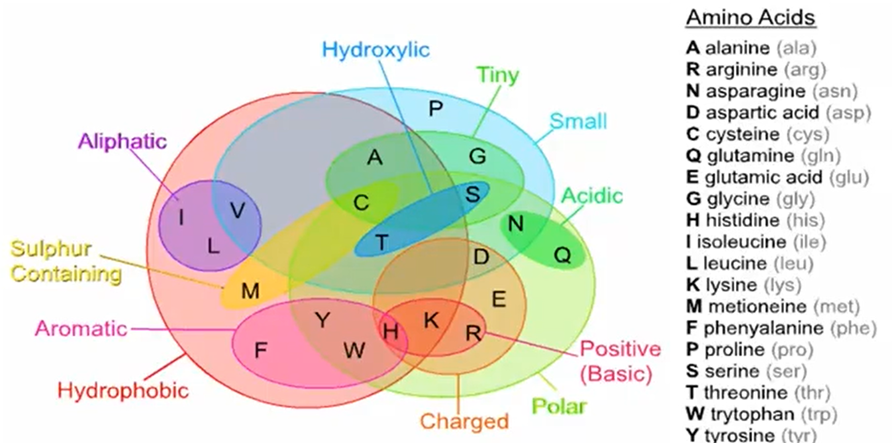
根据氨基酸的性质相似程度如上图,我们对任意两个氨基酸之间的对齐进行打分,如下图:

对于任意两个氨基酸序列,可以通过补空位,左右移动位置等等,使得匹配的全局得分达到最高,此时我们就得到了两条氨基酸序列的对齐。同样的,我们可以对多条氨基酸进行对齐。现在我们拥有很多的MSA搜索工具,基于一些局部匹配算法,我们输入一条想要分析的氨基酸序列,可以很快地在氨基酸序列库中搜到相似的氨基酸序列并进行对齐。
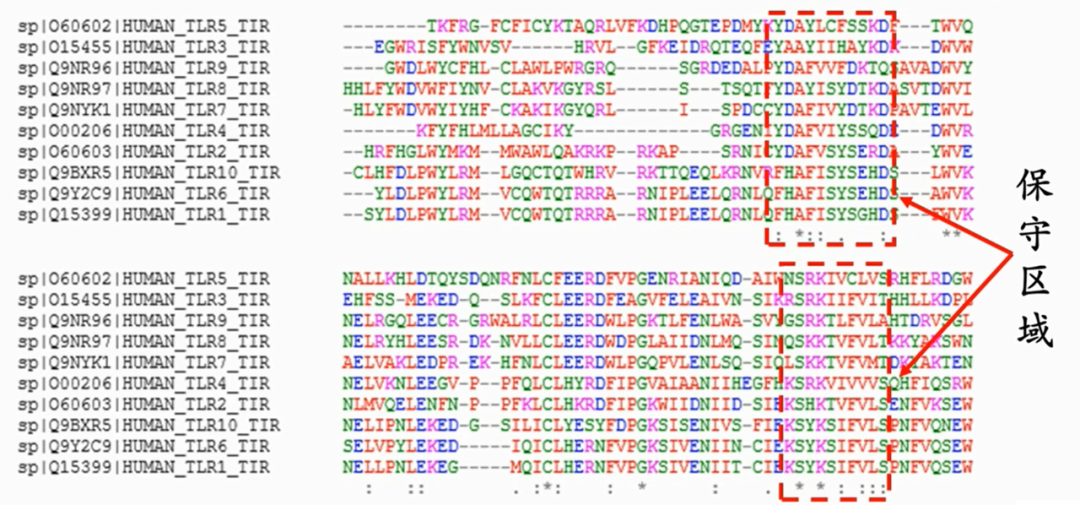
那么我们做多序列对齐MSA有什么用呢?主要目的是通过共进化分析找到保守区域和其他特征。我们假设找到的相似蛋白质是同一个祖先进化来的,生物体内时常发生突变,如果突变发生的位置在蛋白质的不重要区域,那么很可能对蛋白质的空间结构影响不大,不改变蛋白质的功能也不致病,而如果突变发生在了蛋白质的靶点区域和产生作用的区域,很可能导致蛋白质功能变化而影响生物的存活。因此保守区域即上图中对齐的很好的部分,很可能是蛋白质发生功能最重要的区域,其空间构型稳定;而那些缺失了增加了或者氨基酸变化很大的区域,则有可能是不重要的区域。
需要指出的是,我们想要得到的是同源蛋白质,而氨基酸相似性搜索只能搜到相似的蛋白质,但如果想要分析这些蛋白质是不是同一个祖先进化来的很困难,因此我们往往使用搜索到的相似蛋白质就假设他们都是同源共进化的蛋白质来使用。(实际上,同源的蛋白质可能因为不同的进化方向而很不相似,而非同源的蛋白质也可能趋同进化变的相似。)实际用的时候,相似性90%以上的氨基酸序列的MSA没什么意义,因为太像了,留一条就够了;相似性30%以下的氨基酸序列MSA也没什么意义,因为太不像了很可能不是同源的。
CASP
Critical Assessment of protein Structure Prediction (CASP),蛋白质结构预测技术的关键测试,是自1994年以来每两年进行一次的全球范围内的蛋白质结构预测竞赛,目的是更好预测和破解蛋白质三维结构。每两年一次的比赛中,还未公开发表论文的刚被测出来三维结构的蛋白质被用来当做测试目标,进行公平的三维结构的预测。2022年又要举办啦,感兴趣的小伙伴们冲啊!
为啥深度学习研究蛋白质结构很重要
1)分析蛋白质功能。蛋白质的三维结构确定其工作方式与功能。生物的基因通过表达为蛋白质等生物分子来进行各种生命活动。研究蛋白质的功能,首先就要知道它以什么样的构型与其他的配体或者蛋白发生作用。预测了蛋白质的三维结构,我们可以得到其活性区间和靶点。对于给定的结合位点,我们可以识别,设计以及改善其配体。我们也可以进行抗原表位预测等等。特别是对于酶或者其他有催化活性的蛋白质来说,他们活性中心的保守性较高,而外周部分的保守性较低,而我们通常的研究方法就是收集他的突变体,进行每个氨基酸残基改变后对蛋白质功能的影响进行研究,为了指导更好的功能研究,首先要对其结构进行一定程度的预测。
2)制药领域的需求。新药的研制流程包括,首先确定病的成因靶点,即哪个蛋白质/哪个基因出了问题,需要去抑制这个靶点还是去激活这个靶点。然后是针对靶点进行对应小分子药物的设计,比如模拟体内的小分子再修改其功能,修改已有相似药物的功能,根据靶点的三维结构去设计对应结构可以结合上的小分子,或者对已知可合成的小分子进行高通量筛选。之后是对这一步中所有可能的小分子进行缩小范围,比如判断其毒性,跨模型,在体内会残留多久,会不会对其他器官/蛋白质产生损害等等。再往后就进入了实验和临床,从试管和小动物开始,逐步到人体试验,整个过程需要花费10到15年去研发一款新药,花费5到10亿美元或者更多。无论是小分子药物,蛋白质药物,还是疫苗的设计,临床前的设计都可以通过AI的辅助减少大量的时间金钱花费,高效地进行靶点判断,药物筛选,毒性判断等等。
3)通过实验进行蛋白质三维结构的测定成本过高。为了测到一个蛋白质的三维结构,需要耗费科学家很大的经历,花费几个月甚至几年的时间进行实验测量。蛋白质的三维结果可以通过核磁共振、结晶然后X射线、冷冻电镜等。可以结晶的蛋白质只占少数,尤其是膜蛋白这种难溶的蛋白质;核磁共振的精度比较低,分辨度和灵敏度都不足,分子量三万以上的蛋白基本上无法使用核磁共振分析;冷冻电镜由于其精度和覆盖范围称为生物大分子结构解析的主要方法,近些年通过冷冻电镜测出来的高精度蛋白质三维结构数量增加,这也使得我们通过机器学习进行蛋白质三维结构预测在数据量上成为可能。但是使用冷冻电镜的价格很昂贵,所需要的技术门槛很高,往往使用冷冻电镜测量出一个蛋白质的精确三维结构就意味着一篇Nature子刊甚至Nature正刊。而AlphaFold2直接把这个问题给解了,精度达到了和冷冻电镜相似,等于好多篇Nature摞起来,这就是这篇“魔改Transformer”的分量,被圈内的人认为是诺奖级别的工作。
4)理解生命。我们做NLP的在用各种技术去理解自然语言和应用自然语言,而生命这门语言同样值得我们分析。生命的密码被记录在DNA,基因中,基因通过被翻译成蛋白质来表达功能。无论是DNA的序列,RNA的序列,还是蛋白质中氨基酸的序列,都是生命的这门语言,我们可以用NLP里序列分析的技术去解读生命的语言。另外举两个小例子,MSA中,同源的蛋白质的氨基酸有多大差异,就往往代表他们从同一个祖先分离出来之后独立进化了多久,我们可以分析氨基酸序列来追溯进化的历史,找到那些还未被发现的共同祖先;序列建模出三维结构,我们进一步可以建模病毒(大多病毒就是DNA/RNA外面罩一个蛋白质壳子),病毒已经介于生命和非生命之间了,而且计算出其生命结构和运行规律看起来已经不远了。如果再进一步如果有一天我们通过人工智能建模出了生命,这事就简直太帅了。
以上各个需求需要生物科学家耗费巨大的时间和精力才能往前推进一点,而AlphaFold2的精准度和速度,使得上面各个生物学进展都可以得到巨大的提速和辅助,并且开启了AI科学家进军生物大分子和探索生命的巨幕,可以预见到,将来会有更多深度学习探索生命的突破性进展,因为AlphaFold2证明了,使用深度学习去解决如此困难的3D空间预测都是可行的。因此,它的贡献度足以顶上很多篇Nature/Science,被称作诺奖级别的工作也不为过。
Related Work
我们先简单的讲一下之前的蛋白质三维结构预测是怎么预测的。首先要知道的一点就是,仅仅通过一串氨基酸序列,就计算出蛋白质里每个原子的三维空间坐标,实在是太难了。因此,MSA,检索模板等技术往往作为辅助手段使用。1)同源建模法。对于给定的氨基酸序列,检索相同蛋白质家族中已知结构的模板,然后做MSA分析,很相同的部分保留模板结构,不相似的部分再根据能量方程(根据物理学知识,势能越低结构越稳定)修改。2)穿线法。已知的蛋白质结构约十万个,不同结构拓扑的仅有1393个,很少有找不到相同结构拓扑的。在已知序列的各个拓扑结构中,使用能量方程,对给定氨基酸序列选取最低最稳定的拓扑结构当模板。3)从头计算法,如果前两种做出来的结构都很差,找不到能用的模板,就可以根据蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态的原则,直接计算最低自由能的结构。4)综合法。把给定的氨基酸序列分成一个一个的片段,每个片段使用上面三种方案中最好的方案,然后再拼回来。对于这些传统的计算方法,由于需要在计算能量方程等内容时使用各种近似和假设来降低计算量,所以虽然之前结果还能看,但是Alphafold一代出来之后,就被黑盒的魔法打败了。所以,接下来我们看一下AlphaFold一代:
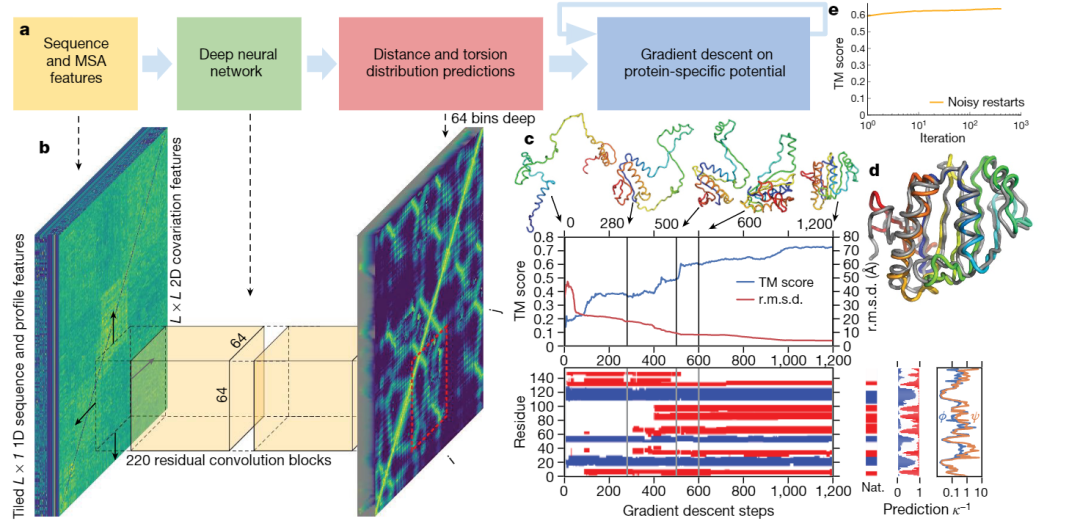
AlphaFold1中,输入是MSA feature,这个MSA里拿出来的特征是使用生物学家的传统算法得到的。神经网络的输出是氨基酸两两间的距离;这个神经网络的输出很好理解,因为三维结构里每个原子的坐标是可以随意旋转和平移的,但是无论如何平移旋转,任意两个氨基酸直接的距离是确定的。(AlphaFold1的源码我读的别人pytorch写的相似的而不是原始论文的代码,此段大家看个意思和大概就行,不要当做完全正确的,想要一步理解的读完大意之后可以自己去调研细节)神经网络预测出来两两氨基酸之间的距离,而我们最终需要的是氨基酸内部所有原子的距离,因此需要上采样到backbone主链原子间的距离。然后拿到backbone原子距离之后,再喂给能量方程的程序包去补足侧链上原子的距离,然后反复迭代优化势能函数去计算出每个原子的坐标。
我们可以看到,AlphaFold1中,并不是end to end去训练的,喂进来的输入,是传统算法得到的MSA特征值,预测的输出是两两残基间的距离而非最终的三维坐标,运算过程中大量使用生物学家提供的计算程序包。即使如此,在CASP13中,仍然领先其他模型很多,开启了AI进军生物大分子界的序幕。后来,CASP14里的AlphaFold2,则采用的完整的end to end训练,使用了Transformer,比赛结束后Deep Mind给出了如下的概念图,但是一直没用公布细节:
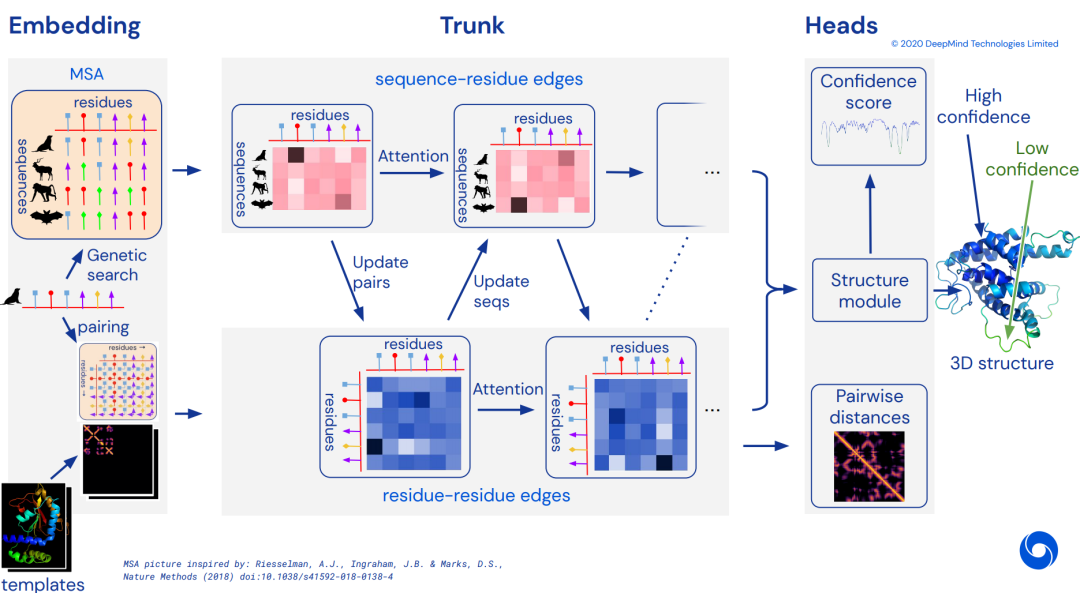
可以看到,AlphaFold2里使用目标氨基酸序列、MSA、模板作为输入,直接end to end的预测了目标的三维结构,使用了Transformer进行预训练。在等待AlphaFold2公布论文细节的过程中,若干使用Transformer在氨基酸序列上进行预训练的工作被开展,比如ESM,MSA Transformer等等,他们在氨基酸序列上,进行BERT、XL-NET、BART等等预训练任务,然后进行二级、三级结构预测、蛋白质性能预测等等任务的finetune。他们往往报了无监督的二级结构预测结果,方法是在预训练后最后一层的attention分布上加一个线性层,使用几个蛋白质去训练这个线性层,然后预测,本质上是few shot。其中,比较亮眼的一个工作是MSA Transformer,效果突出,在AlphaFold2开源之前,被认为是AlphaFold2里的MSA部分的一个很好猜测拟合,如图:
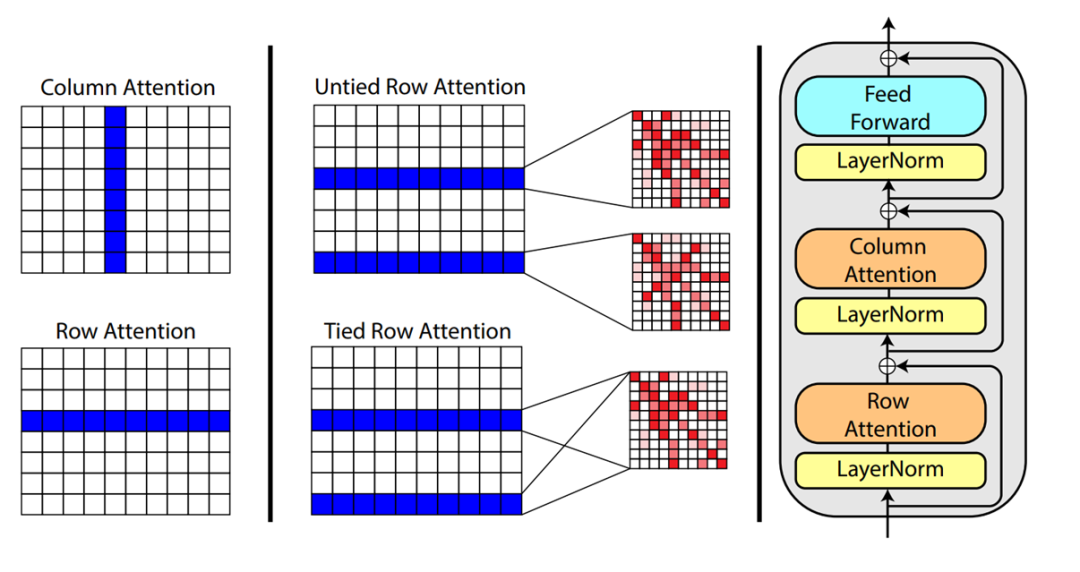
在MSA Transformer里,MSA被作为输入,每层的注意力层被拆分为了横向attention和纵向attention。其中,横向attention就是每个氨基酸序列里的self-attention,纵向attention是相同位置的去看其他氨基酸序列里是否被替换了氨基酸还是大家都相同。它比较神奇的一个结论是,最好的下游任务结果出现在,横向的attention所有的氨基酸序列共享attention分布,即,使用整个MSA的横向attention平均值作为每个蛋白质的attention。这就意味着,MSA Transformer的结论可以被认为是,单个氨基酸序列的信息表达太差了(比如之前的工作),还不如使用整个MSA的平均值去表达更通用的信息。(然而事实真的如此吗?很可能是因为之前对单个氨基酸序列的建模能力太差而已,更好的结构应该是MSA得到更多的信息和特征,再用我们想要的那条特定氨基酸序列进一步优化修改,比如我们将要看到的AlphaFold2。)同期还有其他的三维结构预测文章,但是效果都比不上AlphaFold2,暂不介绍。与AlphaFold2同一天放出来的另外几个工作效果很好,与AlphaFold2性能相似甚至更好,时间所限,还来不及阅读(RoseTTAFold等)。
Alphafold2
算法简述
根据上面介绍的基础知识和相关工作,我们感叹这个工作确实难度很高,也非常好奇AlphaFold2里究竟如何使用MSA信息,如何又增强突出出来当前自己这条氨基酸序列的能力,如何使用的模板信息,又是怎样超直接end to end训练的三维位置坐标的。过去的半年里,很多小伙伴等不及了,甚至猜着模型结构去开源AlphaFold2的猜测代码[3],甚至获得了500多个星标。如今,我们终于盼来了 真正的开源代码[4] ,在写这篇文章时(开源后一天的周六),已经获得了超过3千的星标。先再次回忆一下之前放出来的结构图:
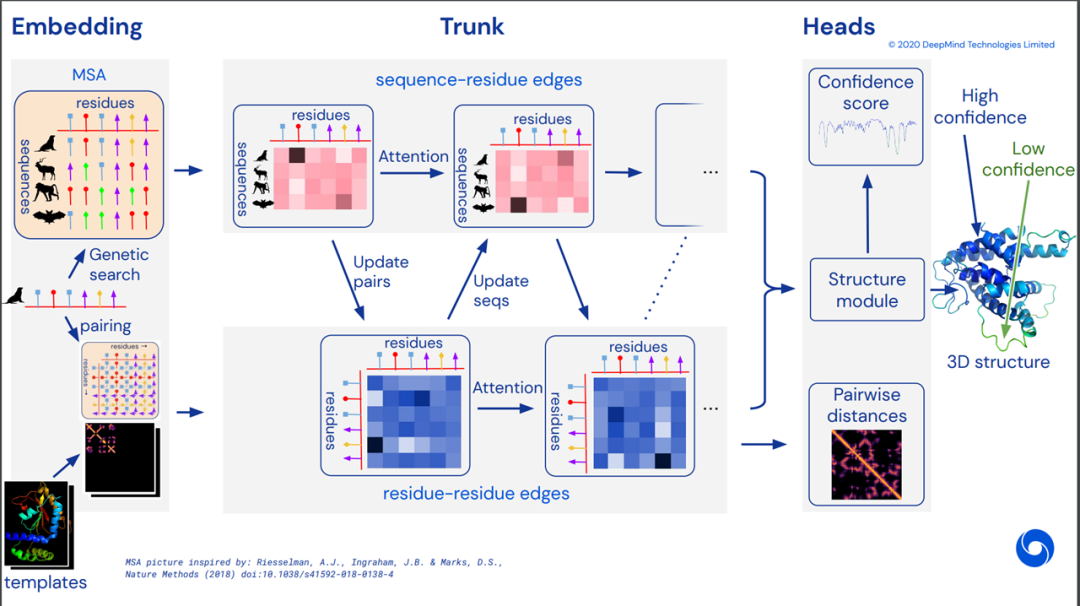
然后我们再看一下这次刚刚放出来论文里的结构图:
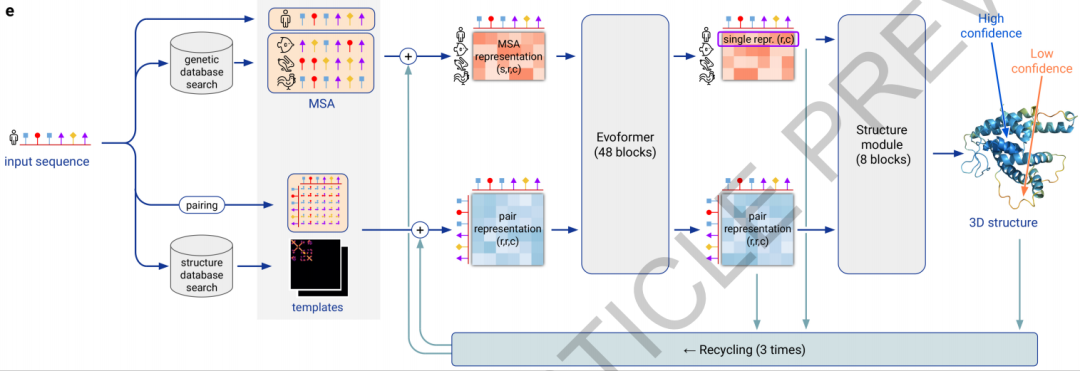
时间匆忙,16页的正文,62页的附录算法,仍然是细节来不及看,我们大体的过一过AlphaFold2算法,反正也不是做这个的,我们NLP的小伙伴一起学习学习思想就可以了~Alphafold2提出了一个新的结构去同时嵌入MSA和残基-残基对儿的特征(没错,不是每个残基一个隐状态,是每个残基之间都有一个隐状态来描述他俩之间的关系,我们把每对儿残基之间的隐状态叫做pairwise features),新的输出表示去确保准确的端到端训练,以及新的辅助loss。此外,在finetune训练之前,AlphaFold2首先预训练了一把,在MSA上使用BERT任务遮盖住一些氨基酸再还原回来,此外还使用自蒸馏,自估计的loss去自监督学习(先用训好的模型在只有氨基酸序列的数据上生成预测结果,然后只保留高确信度的,然后使用这个数据预训练,在训练时把输入加上更强的drop out和mask,来增大学习难度,去预测完整信息时高确信度的结果)。结构由两部分组成,Evoformer和结构模块(Structure Module)。Evoformer输入MSA,模板,自己的氨基酸序列,输出MSA信息和残基-残基对关系(刚刚提到的pairwise features)建模。结构模块中,丢掉MSA中的其他氨基酸序列,只保留目标的那一条,然后再加上pairwise features,去计算更新backbone frames,预测所有氨基酸的方位和距离,肽键的长度和角度,氨基酸内部的扭转角度等。
Evoformer即进化former,进化魔改Transformer,用来计算MSA和pairwise features。开源之前我们猜测模板是很重要的一个信息,结果发现匹配到的模板信息仅仅通过一些特征提取的函数被塞进了MSA里面当成给目标氨基酸序列做更新时参考使用的数据。具体结构如下图,输入MSA和pairwise features,通过很多注意力层,最终输出MSA和pairwise faetures
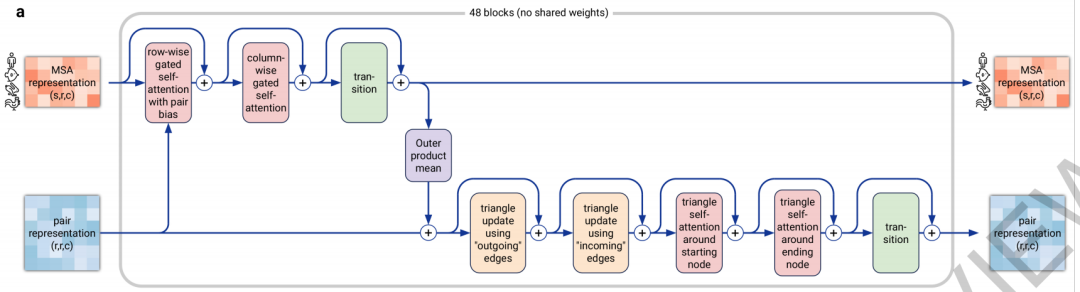
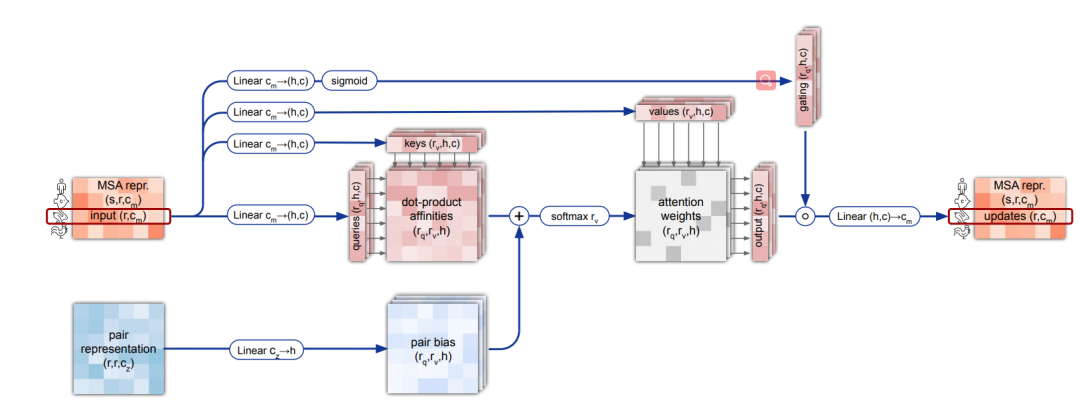
我们看到具体的结构,之前MSA Transformer猜测的大体不差,有横向attention去往自己当前氨基酸序列的所有位置打attention,有纵向的attention去看其他氨基酸序列的相同位置稳定性和突变有多少,只是在横向attention时,加了使用pairwise features作为 后attention上的一个bias:
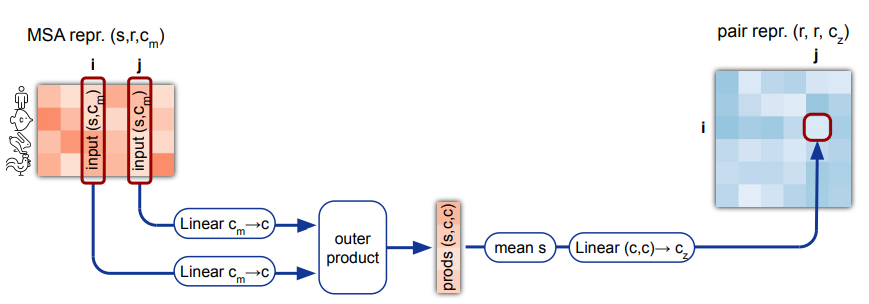
MSA经过横向和纵向的attention,最后还有一个transition层,其实就是两层的MLP,中间塞一个relu。transition层的输出结果,通过计算外积,然后求平均,去计算出pairwise features:
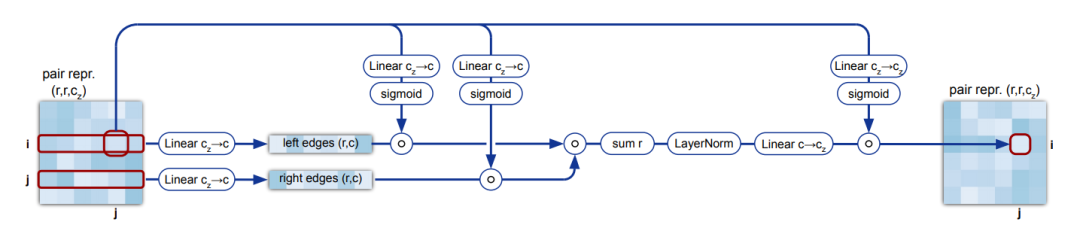
之后是pairwise features的更新,称为三角乘法更新(Triangular multiplicative update)。其motivation是说,这个矩阵描述的是任意两个残基之间的距离等关系,距离关系不是自由的,应该满足比如三角不等式,相邻的三个边应该满足两边之和大于第三边,所以这里的pairwise feature的更新使用了所谓三角乘法更新,对于每个边,都会接收到和他组成三角形的任意两个其他边带来的更新:
之后是两层三角自注意力层(Triangular self-attention):
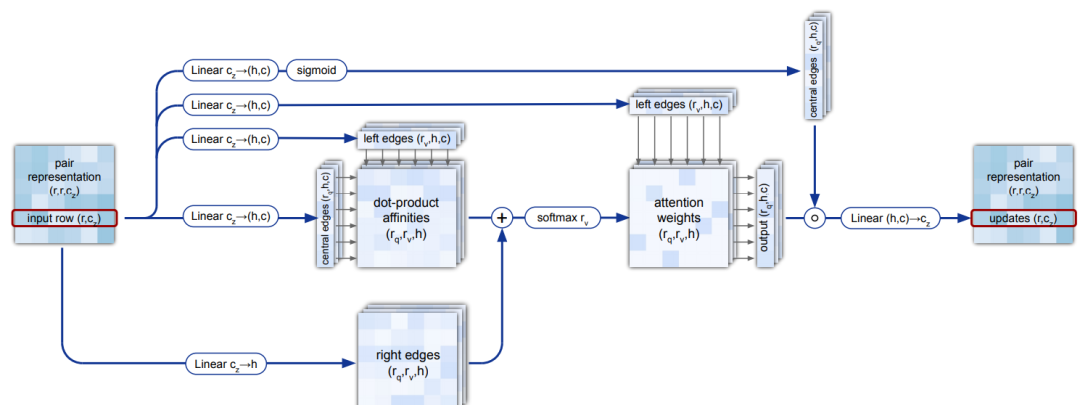
结构模块(Structure module)结构模块使用MSA中的第一条而丢弃剩余部分(即只保留目标的这个氨基酸序列,丢掉检索到的其他MSA,称作single repr),以及计算得到的pair features,以及把所有残基都从坐标原点初始化然后再去计算更新的backbone frames,最终预测出具体的3D原子坐标。在计算中,每一层都去更新single repr和backbone frames(每个残基一个backbone frame,每个backbone frame记录了从局部坐标系到全局坐标系的欧几里得变换),而计算得到的pair features只在更新single repr的attention层中计算成一个bias。(之前的猜想都以为要从pair features上计算得到每对儿残基之间的距离,没想到只是个小小的配角)
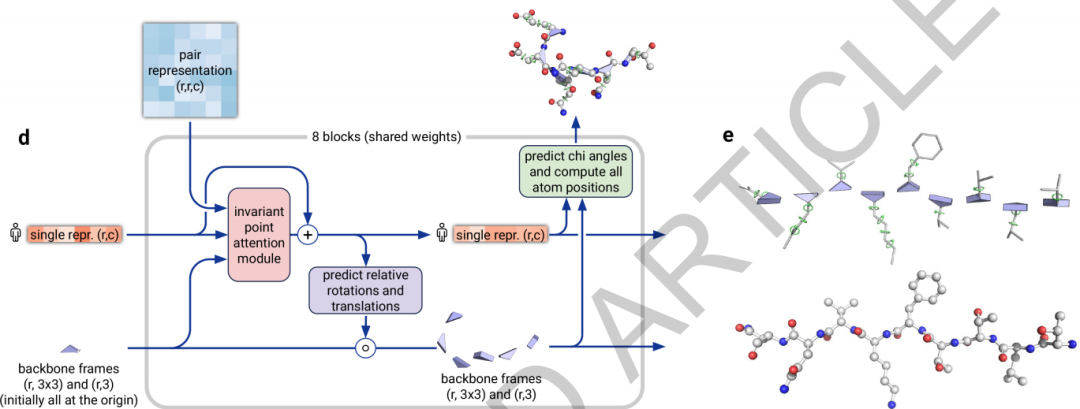
然后我们逐层看结构模块在算什么。首先,这个backbone frames使用“黑洞初始化”,这个黑洞初始化把所有的残基都放在全局坐标的坐标原点,然后在后面的每一层计算推走多远。pair features只用来在第一个attention子层中计算bias辅助更新single repr,他们永远不变。在Invariant Point Attention Module子层中,更新single repr,在Predict Relative Rotation and Translation子层中,single repr被映射为具体的update frames去更新backbone frames。对于每个残基,我们预测它和相邻残基之间肽键的角度和距离,这个角度和距离是自由的;而为了确定每个原子的坐标,残基内部也有若干扭转角度的参数,残基内部确定原子位置仅由这些扭转自由度确定。
效果展示
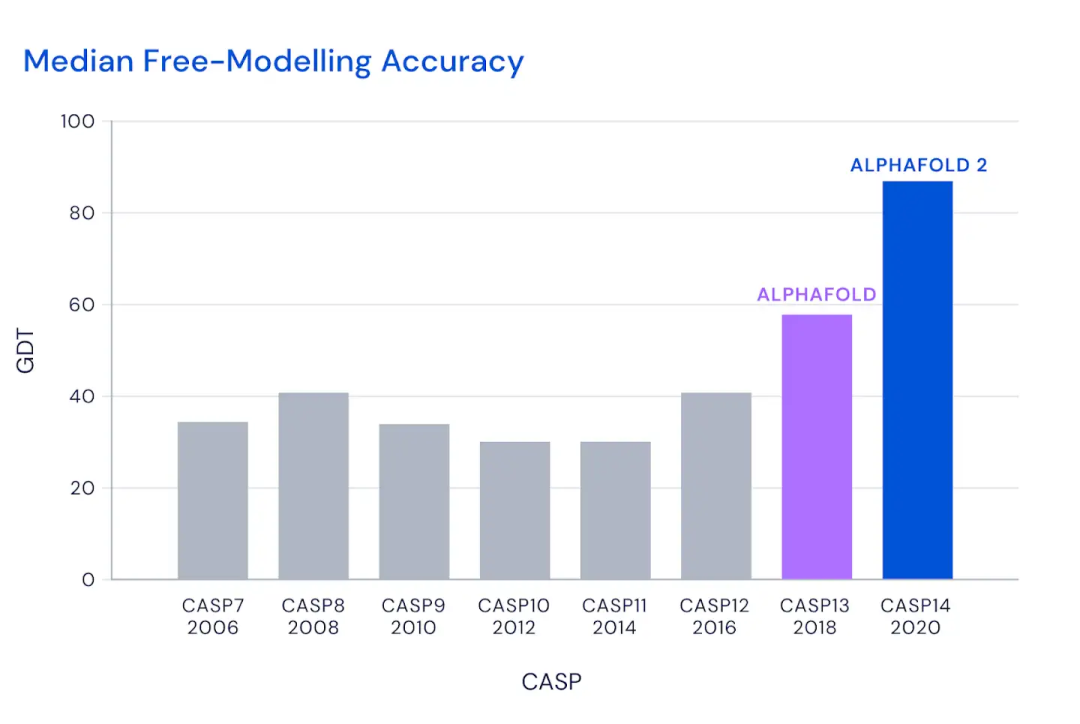
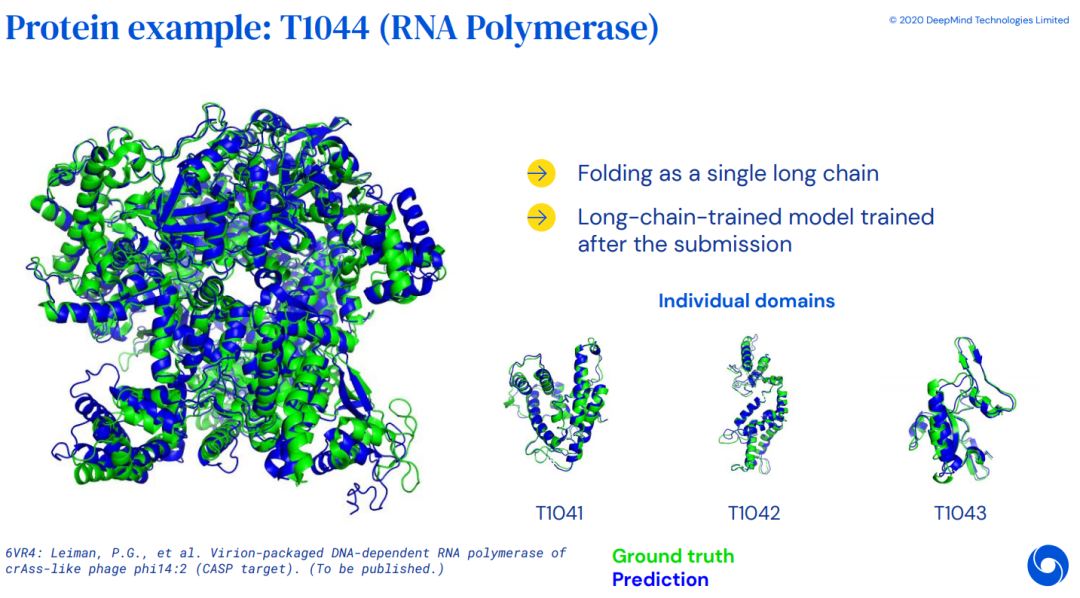
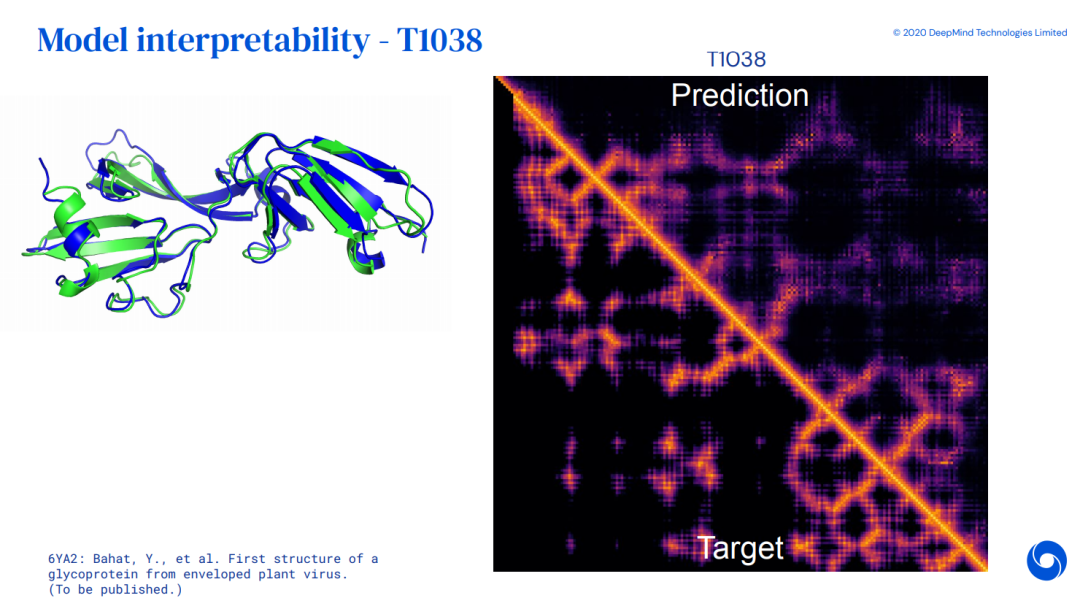
总结和展望
本文主要介绍了蛋白质三维结构预测的背景知识和相关概念,让大家感性和部分理性上理解一下AlphaFold2很厉害。如果后续有比较多的人对AlphaFold2的具体细节和技术感兴趣,那么可以继续更一个详解的讲解;不过我们应该大部分读者都是做NLP的,了解了隔壁领域的大概意思学习其思想就可以了,不是那么着急的可以等专门搞生物的人的详解文章。未来不出意料的话,会有更多的研究员和计算资源被投入到生物大分子领域,人工智能发展和生物、医疗、制药,都会在未来的几年迎来一波技术的小爆发。
萌屋作者:炼丹学徒
在微软搬砖的联培博士在读生,擅长烹饪和摸鱼,被迫掌握丰富的增肥和减肥经验。祝大家吃好喝好,减肥成功。
作品推荐
 萌屋作者:炼丹学徒
萌屋作者:炼丹学徒
后台回复关键词【入群】
加入卖萌屋NLP/IR/Rec与求职讨论群
后台回复关键词【顶会】
获取ACL、CIKM等各大顶会论文集!
![]()
[1].论文算法附录:https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03819-2/MediaObjects/41586_2021_3819_MOESM1_ESM.pdf
[2].博客:https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology
[3].猜测代码: https://github.com/lucidrains/alphafold2
[4].真正的开源代码: https://github.com/deepmind/alphafold
 后台回复关键词【
后台回复关键词【