图神经网络(GNNs)作为深度学习中具备拓扑/结构感知能力的模型,已成为人工智能辅助药物发现(AIDD)中的强大工具。通过直接作用于分子图,GNNs 提供了一种直观且富有表现力的框架,用于学习类药分子复杂的拓扑和几何特征,从而巩固了其在现代分子建模中的重要地位。本文综述了 GNN 在药物发现中的方法基础与代表性应用,涵盖了分子性质预测、虚拟筛选、分子生成、生物医学知识图谱构建和合成路径规划等任务。我们重点介绍了一些最新的方法进展,包括几何图神经网络、可解释模型、不确定性量化、可扩展图结构以及图生成框架。同时,文章还探讨了这些模型如何与现代深度学习方法融合,如自监督学习、多任务学习、元学习与预训练。贯穿全文,我们也指出了 GNN 在实际药物研发流程中面临的挑战与方法瓶颈,并在最后讨论了未来的发展方向。 关键词:图神经网络;分子图;深度学习;人工智能辅助药物发现;分子建模


1.1 图在药物发现中的自然表示形式
药物发现是一项资源密集型且耗时漫长的工作。据估计,将一种新药推向市场通常需要超过 20 亿美元的投入,并且研发周期往往超过十年【1】。这一过程中的高成本与高风险限制了能够通过药物干预有效治疗的疾病范围。因此,任何能够带来方法论突破、尤其是重塑基础范式的进展,都会引起学术界和工业界的广泛关注【2】。随着时间推移,药物发现已从早期依赖偶然发现的方法,逐步演进为高通量筛选(HTS),再到更近的基于机制的理性设计,向着更高的可控性与特异性不断推进【3】。
近年来,人工智能(AI)的快速发展为药物发现注入了新的动力,加速了从基于规则的启发式策略向数据驱动方法的转变。2024 年诺贝尔化学奖授予了蛋白质设计方面的突破性成果,进一步凸显了将 AI 融入药物发现常规流程的变革潜力【4】。除了算法上的进展,结构生物学与多组学等领域高质量实验数据的日益丰富,也为 AI 的应用奠定了坚实的数据基础。像蛋白质数据库(PDB)【5】和癌症基因组图谱(TCGA)【6】这样的数据库提供了大规模的结构、转录组与蛋白质组数据,对数据驱动建模至关重要。在这一背景下,人工智能辅助药物发现(AIDD)正逐步形成一个系统级范式,涵盖靶点识别、先导化合物筛选、候选优化和早期风险规避等环节【7】。AIDD 主要应用于药物研发的早期阶段,其核心目标是寻找并优化候选分子的特异性与可开发性,同时最大程度降低毒性与临床试验失败风险。这些进展共同推动了药物研发流程的提效降本【8】。
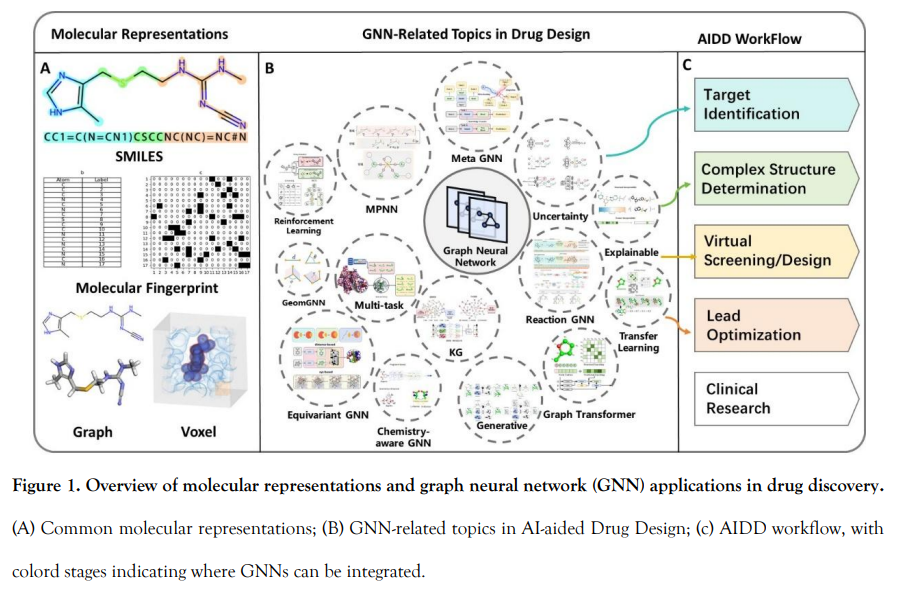
要充分释放 AI 在药物发现中的潜力,必须首先将分子结构编码为机器可读的表示形式。为此,研究者发展了多种分子表示策略,包括基于字符串的线性化方法(如 SMILES【9】与 SELFIES【10】)、分子指纹【11】,以及基于图和三维体素网格的表示方法【12】,如图 1A 所示。当这些表示与深度神经网络(DNNs)、卷积神经网络(CNNs)【13】以及 Transformer 架构【14】结合时,在构效关系(QSAR)建模和打分函数开发等任务中表现出了良好效果【15】。然而,每种表示方法都有其内在的局限性。指纹和描述符依赖专家定义的特征,往往由于对预定义统计或物理化学模式的依赖而表现力受限【16】。基于字符串的方法将分子结构线性化,而从本体论角度来看,分子本质上是图结构对象:原子构成图的节点,化学键对应边,此外还存在描述其构象几何的三维坐标。
这一形式天然契合图神经网络(GNNs)的建模范式。GNN 通过局部消息传递机制捕捉原子之间的配位模式,并逐层聚合,编码为分子层级的语义信息。通过引入等变或不变算子(如 SO(3)-等变 GNN),GNN 能以对称感知的方式建模立体化学、电荷分布和空间构型。相比传统“扁平化”的表示方法,图结构表示具有三大核心优势:(i)物理一致性,保留拓扑和几何信息;(ii)表示的通用性,可广泛应用于性质预测、生成建模和药理剖析等任务;(iii)增强的可解释性,可通过子图注意力、边归因和结构显著性图实现结构理解。
因此,图表示正在逐渐成为分子建模中的主流。GNN 已被制药巨头(如 Roche、Novartis)、AI 生物技术初创公司(如 BenevolentAI、Insilico Medicine)及开放数据集支持的研究社区(如 OGB-Mol【18】与 GEOM【19】)广泛采纳。这些发展不仅反映了 GNN 在 AIDD 中的核心地位,也表明了对面向多样生物医学任务的专用 GNN 架构日益增长的需求。
1.2 本综述的范围与结构
本综述旨在系统梳理现代 AIDD 背景下图神经网络(GNN)的方法基础与实践挑战。与以往集中于具体任务或经验对比的综述不同,本文采用以模型为中心的视角,探讨 GNN 的概念基础及其在多样化分子设计任务中的作用,如图 1B 所示。我们重点关注以下几个前沿研究方向:大规模 GNN 架构、几何深度学习、可解释性、不确定性量化、图生成建模以及强化学习(RL)。此外,本文还讨论了 GNN 架构如何融合当代深度学习范式,包括自监督学习、多任务学习、预训练与元学习,以提升其在真实药物发现场景中的数据效率、泛化能力和鲁棒性。 本文强调了一种融合数学严谨性与化学直觉的统一建模框架,面向从事 AI 药物研究的化学与生物学研究人员,以及致力于开发生化应用模型的计算科学家。通过揭示表面上不同任务之间的底层逻辑与结构关联,我们希望帮助读者建立清晰的概念框架,从而有效地将 GNN 技术应用于多样化的药物发现挑战。 接下来的内容安排如下: * 第 2 节:介绍 GNN 的方法论基础,包括:(2.1)图学习核心原理(频域与空域框架);(2.2)面向三维分子系统的对称感知设计原则(不变与等变模型);(2.3)大规模 GNN 的可扩展性挑战(如过平滑、过压缩与预训练策略);(2.4)图生成与结构编辑方法;(2.5)图编辑与强化学习技术。 * 第 3 节:探讨分子性质预测,包括 GNN 的可解释性与不确定性量化。 * 第 4 节:聚焦虚拟筛选任务,包括结合位点预测、蛋白-配体对接、亲和力打分与结构预测模型(如 AlphaFold)的进展。 * 第 5 节:介绍 GNN 驱动的分子生成与强化学习方法,用于分子设计与优化。 * 第 6 节:讨论 GNN 在知识图谱构建与推理中的应用,尤其针对生物医学实体。 * 第 7 节:关注化学合成建模,包括逆合成、反应条件预测与基于分子生成的合成路径规划。
这些章节共同构建了理论模型与实际应用之间的双向桥梁,推动基础方法在真实药物发现流程中的有效部署。