


**导读:**本次分享的主题是几何图神经网络在药物发现中的应用。主要包括以下几部分内容:
百度生物计算平台简介 * 基于3D空间结构的药物亲和力预测 * 基于几何图神经网络的小分子性质预测
分享嘉宾|周景博博士 百度研究院 资深研究员 编辑整理|王龙飞 出品平台|DataFunTalk
01
百度生物平台简介****1. 生物医药行业面临的挑战生物计算从2020年开始成为一个非常热门的方向。在过去半个世纪的时间里,生物经济市场规模不断扩大,其中最重要的投入就是制药业。但是我们也看到制药的投入产出比是不断下降的,因为靶点和小分子都已经被进行了充分的挖掘,近10年每十亿美金投入产出的药物数量发生了显著的下降。从巨大的化合物空间筛选出一个潜在的药物分子出来,是机器学习最有可能提升的阶段。直接做计算的仿真或者化学生物实验,都面临耗时长,成本高的问题。如何用机器学习的模型,来更快的找到潜在的小分子化合物,就能够降低临床前的研发成本,从而降低整个生物制药的投入产出比。2. 螺旋桨PaddleHelix生物计算平台

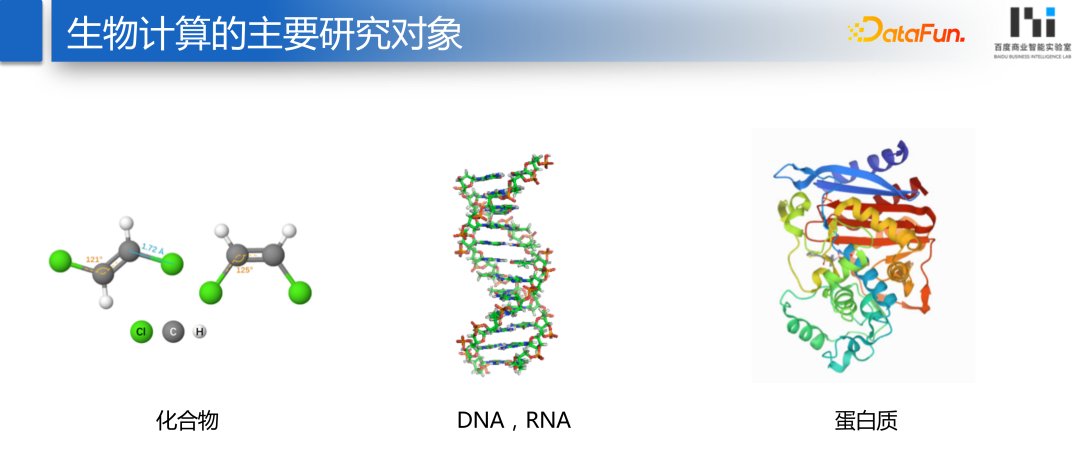
- 化合物
第一类就是化合物,也就是小分子药物。 DNA、RNA
第二类是在基因层面,DNA或者RNA。
蛋白质
第三类就是蛋白质,涉及到宏观的大分子的层面。它的分子数量达到上万甚至几十万,远大于化合物的分子量。这三种数据对于做机器学习来说并不是很陌生,不管是化合物、DNA、RNA还是蛋白质,都可以表示成序列的形式。但是生物数据的几何构型也发挥了非常大的作用,比如像小分子化合物,它们的几何构型可能是类似的,但是由于手性对称、顺式/反式对称等都会导致化合物呈现不同的属性。对于蛋白质也是,蛋白质的功能也是通过几何构型来体现的。所以,我们用机器学习的方式来做生物制药的研究,就要考虑用机器学习的模型更好地建模生物学的数据。4. GNNs with geometric and topological informationGraph Convolution是最流行的机器学习算子,它相对于Convolution最大的改进就是在图结构上进行卷积操作。但是Graph Convolution有一个明显的问题就是几何结构不敏感。Graph Convolution主要的考虑的是图的拓扑信息,比如说这两个节点交换位置,GNN会认为输入是一样的。这对建模分子会产生非常严重的问题。两个分子有不同的构型,不同的构型会产生不同的性质,如果我们认为他们是相同的输入,对应模型的表现会产生非常大的影响。
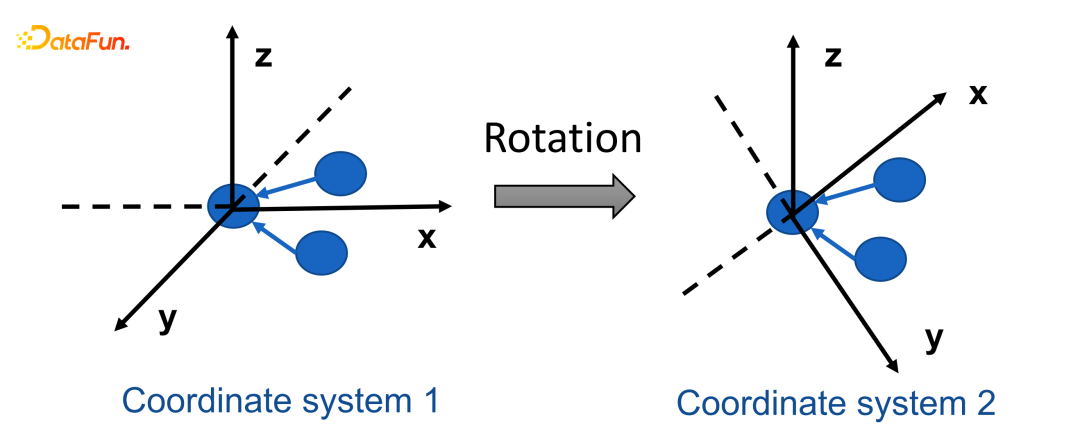
如上图例子,以一个分子作为坐标原点,周围还有两个相连接的分子,我们希望这个分子图的结构信息能保留,此外如果我们直接将相关的相对位置信息进行建模,坐标系进行旋转,它们在坐标系中的取值是不一样的,但是整个分子是没有发生任何变化的。所以我们在进行建模的时候直接encoder spatial information是不行的,还需要考虑这种结构不变性的关系。我们要保证,在表征分子空间信息的时候是几何变换无关的,不能因为坐标系的变化导致整个数据输入发生变化。****这个问题的解决方案大概分为两种:****一个是Equivariant Neural Networks(等变神经网络),这两年有非常多的进展,从2018年开始获得了比较多的关注。简单来讲,等变神经网络就要求对Convolution和Transform要求是等价的,先做Transform再做Convolution还是先做Convolution再做Transform,要求取得的结果是一样的,这样就可以保证几何结构不变性。另外一个就是Geometric encoded message passing,即通过Geometric Encoding的方式来提升GNN的Message Passing,想办法encode相关的几何结构信息来提升在生物数据建模方面的表现。我后面的工作主要是在沿着第二个方向(Geometric Encoded Message Passing)来做的。
02
基于3D空间结构的药物亲和力预测参考文献:"Structure-aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity”; KDD 2021.1. Background
- 虚拟筛选
虚拟筛选指的是通过虚拟的方式事先过滤一些小分子。比如,发现一个靶点后,希望有一个药物跟靶点相结合,进而改变后续下游的生理过程,从而使相关疾病能够被克服掉。一个挑战性的问题是,靶点被发现后,如何找到合适的药物。整个药物分子的空间非常巨大,真正做实验非常耗时且成本非常高。如果能用机器学习的方式,在给定靶点的结构和小分子结构之后,预测出它们两个的亲和力的大小,那么可以加快后续的药物实验和临床实验的效率。
- Protein-Ligand Binding Affinity
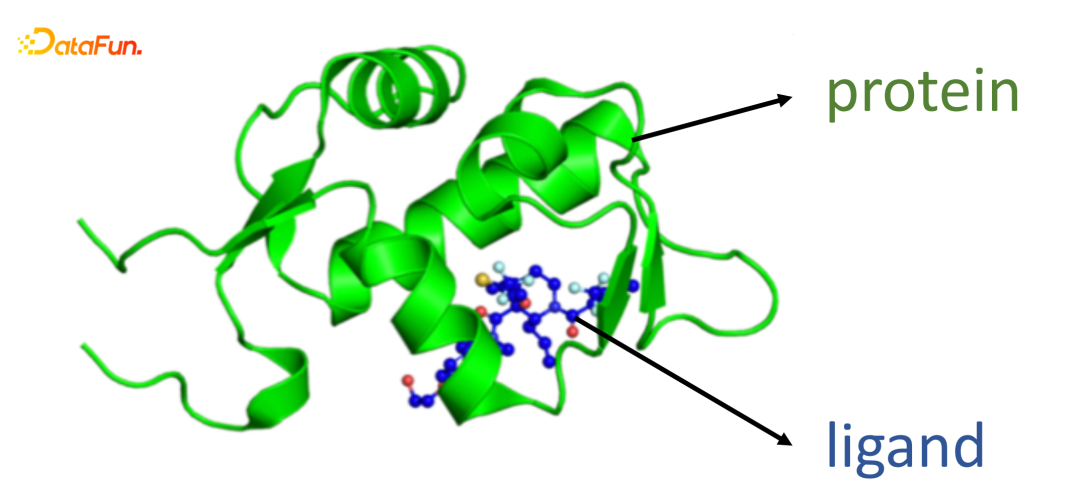
所以,我们这方面的研究目标是,如何预测protein和ligand之间的结合力的强弱。这里相对于其他已有的研究工作,我们着重于利用蛋白质和分子的三维结构信息。
- Structure-based Binding Affinity Prediction
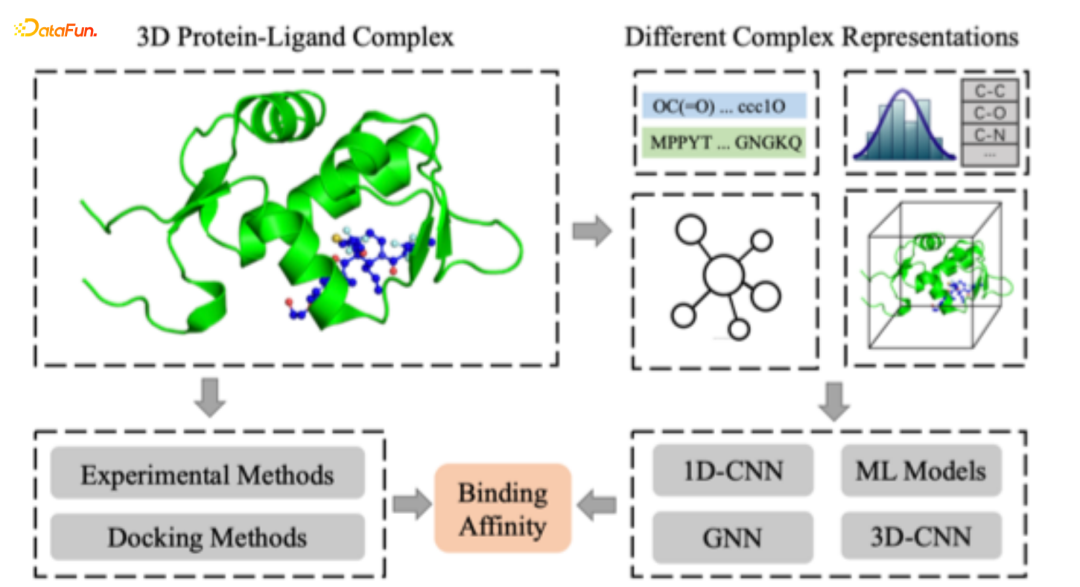
在过去几十年的时间里,亲和力预测也获得了非常多的关注,但是主流的方法可以分为4类:①1D-CNN的方式,按照分子和氨基酸的序列来建模;②按照特征抽取的方式,通过深度学习模型,比如决策树、GBDT、SVM等来预测;③按照3D-CNN的方式,将整个蛋白质口袋和药物结合的位置做切割,利用类似3D grid 的model,像图片处理一样用卷积的方式选表征;④用图神经网络的方式,来提升预测的准确度。
Complex Interaction Graph Construction
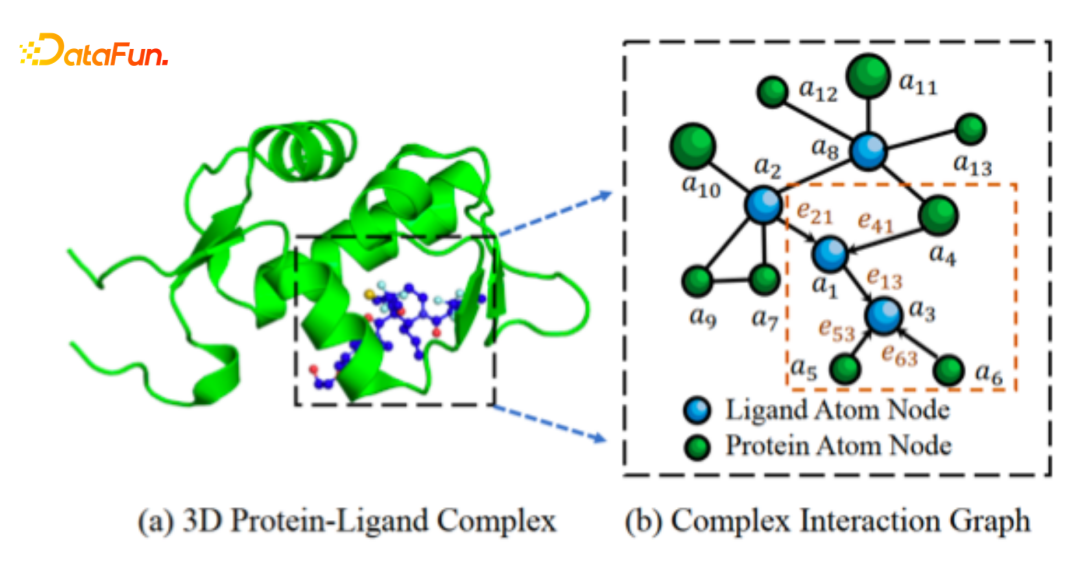
对于GNN的使用,如果想提高准确度,我们主要考虑两个信息,一个是距离信息,需要encode任意两个点之间的相对距离;第二个是角度信息,我们要建模2个原子之间或者3个原子之间形成的键位角。此外,我们也考虑两种不同的键长,一种是共价键,另外一种是非共价键,在原子距离不是很近但也不是很远情况下也存在作用力,主要体现在范德华力。
2. The Proposed Model
Structure-aware Interactive Graph Neural Network (SIGN)
这个是我们的框架。
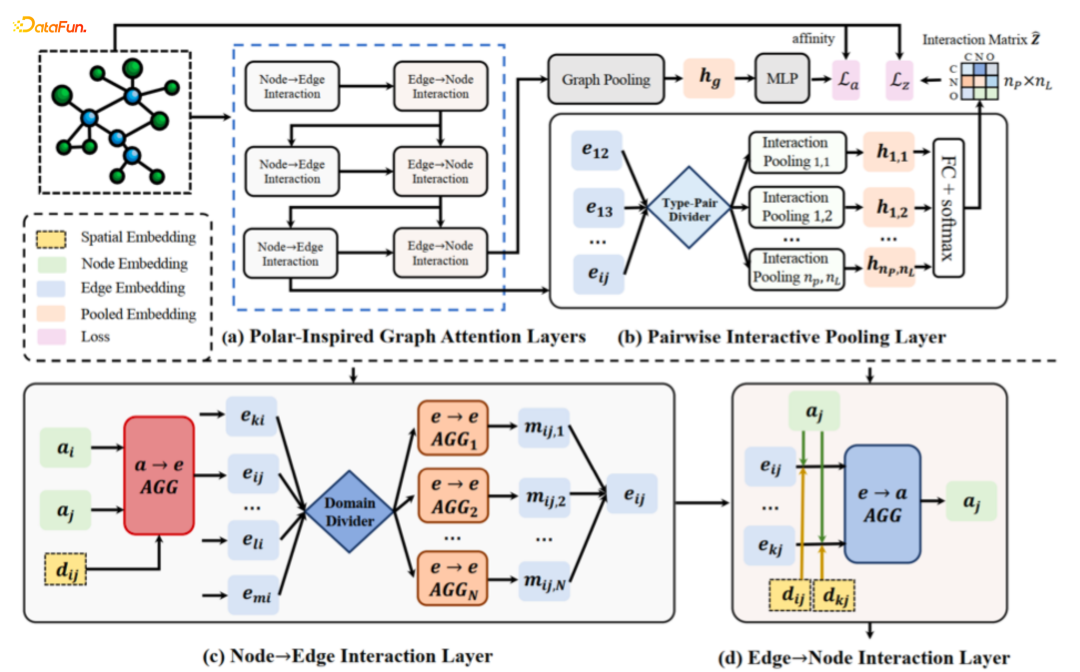
- Polar Coordinate-Inspired Graph Attention
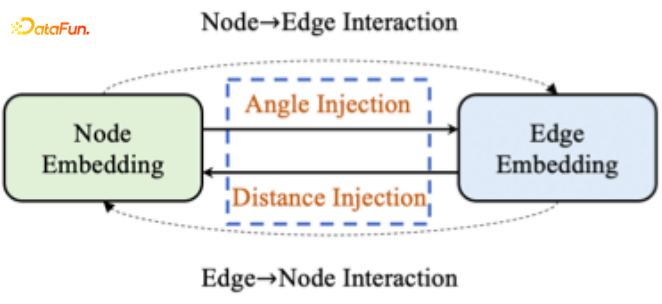
第三个我们还引入了一个方法,就是node-edge的interaction,来提升模型的表现。
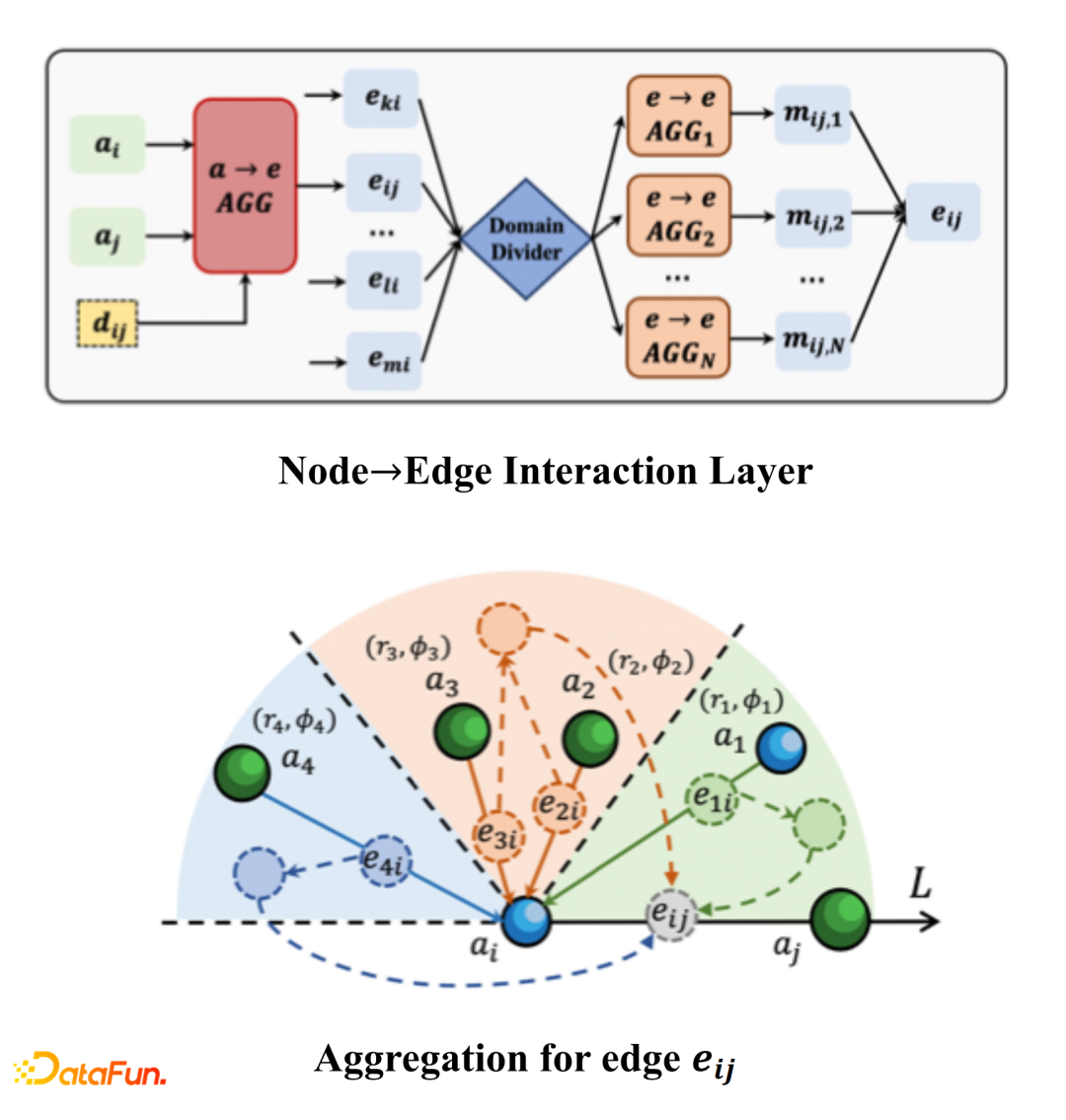
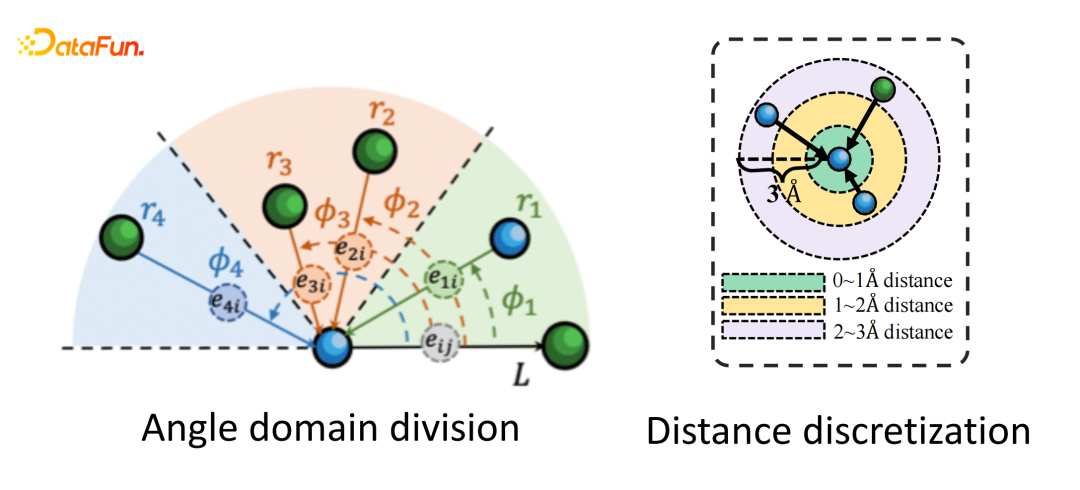
现在简单介绍下如何做角度的离散化。对于一条边ai和aj,我们把它当成中心法线,然后看它在圆锥曲面里和邻居边是什么样子的,这个卷积会定义一个虚拟节点,让所有在圆锥面里的边形成虚拟节点来做卷积,进而在每一个圆锥平面都会定义虚拟节点。最后将虚拟节点进行二次聚合,学习一个global的信息。我们希望在建模的时候,能够将不同的角度信息包含到表征学习的过程中。聚合的过程,首先会在每一个sector来学一个聚合的表征,最后会做一个global的aggregation,进而学习edge的表征。
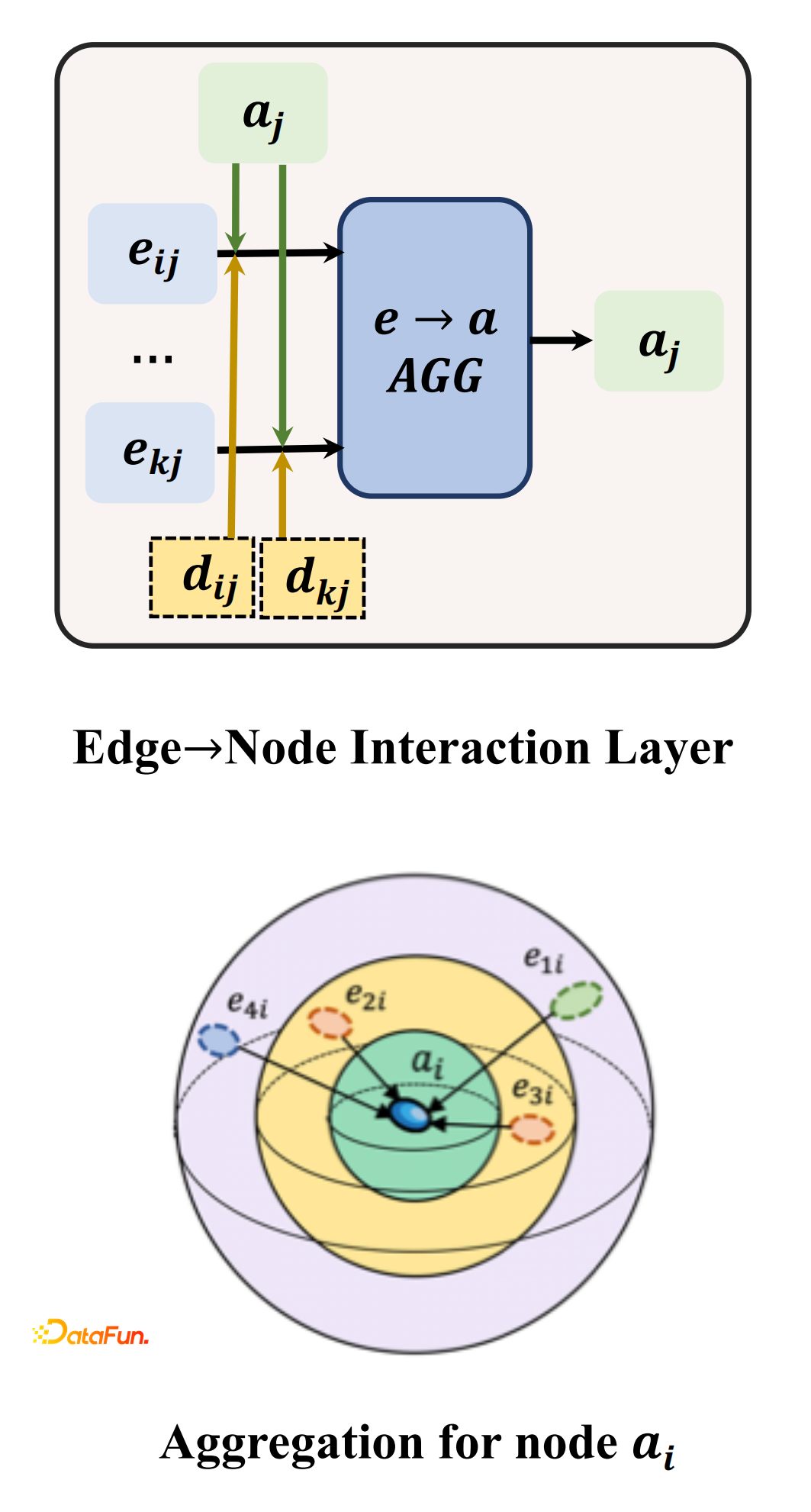
Distance也是类似的,我们会学每个distance,在不同的distance情况下我们认为表征是不一样的,这里我们引入attention机制,按照不同的level和不同的权重聚合到中心节点上,然后将两个表征融合到一起来生成一个节点的表征,做下游的预测任务。
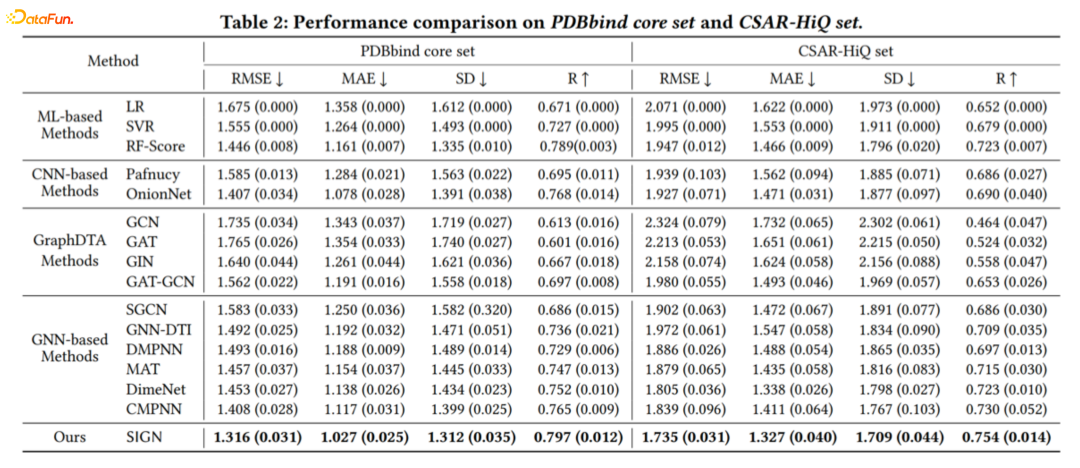
3. Experimental Results这个是我们做的一个实验。
- Datasets
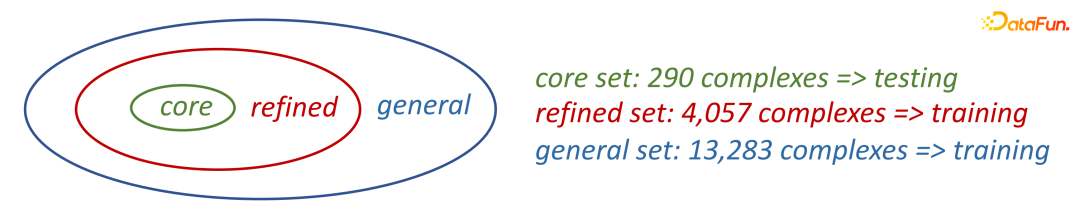
这个数据集PDBbind是一个公开的benchmark,来做预测的,总共有3个set,一个是general set有13283个,refined set有4057个,core set有290个。此外还有另外一个数据集CSAR-HiQ,来做额外附加的验证。
- Baselines

- Comparison with baselines
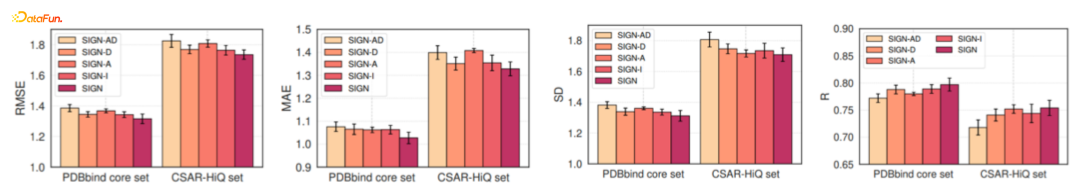
- Impact of Spatial and Interactive Factors
- Molecular Property Prediction
- Graph Representation Learning for Molecules
- Geometrics Structure Learning on Graphs
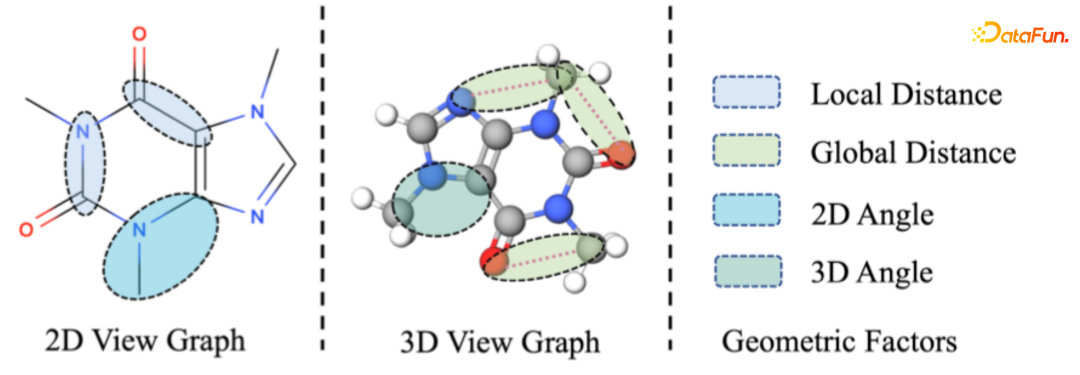
- Contrastive Learning on Graphs
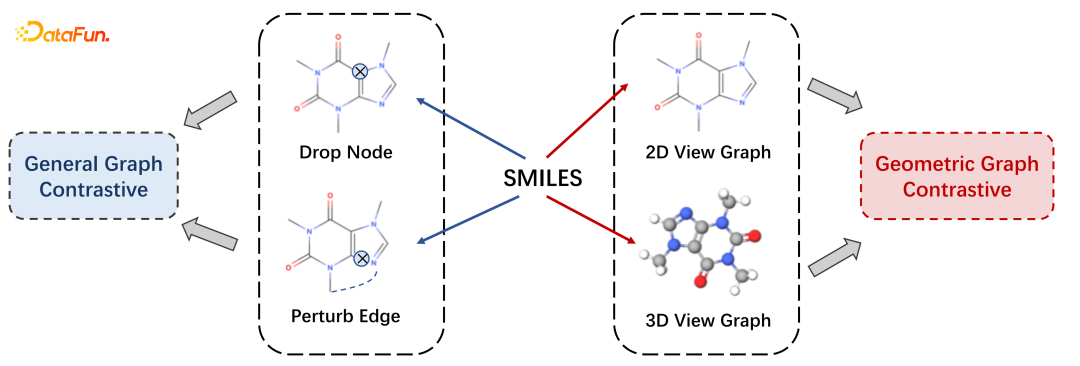
- Geometric Graph Contrastive Learning
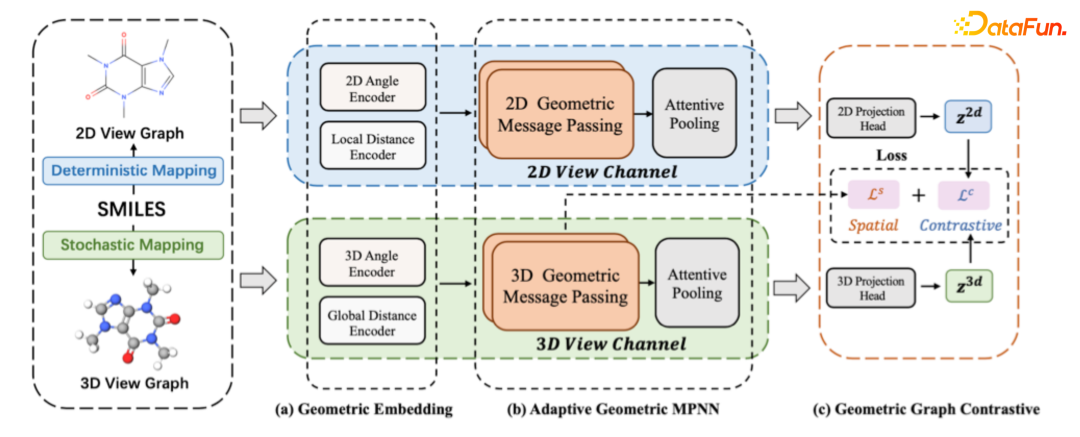
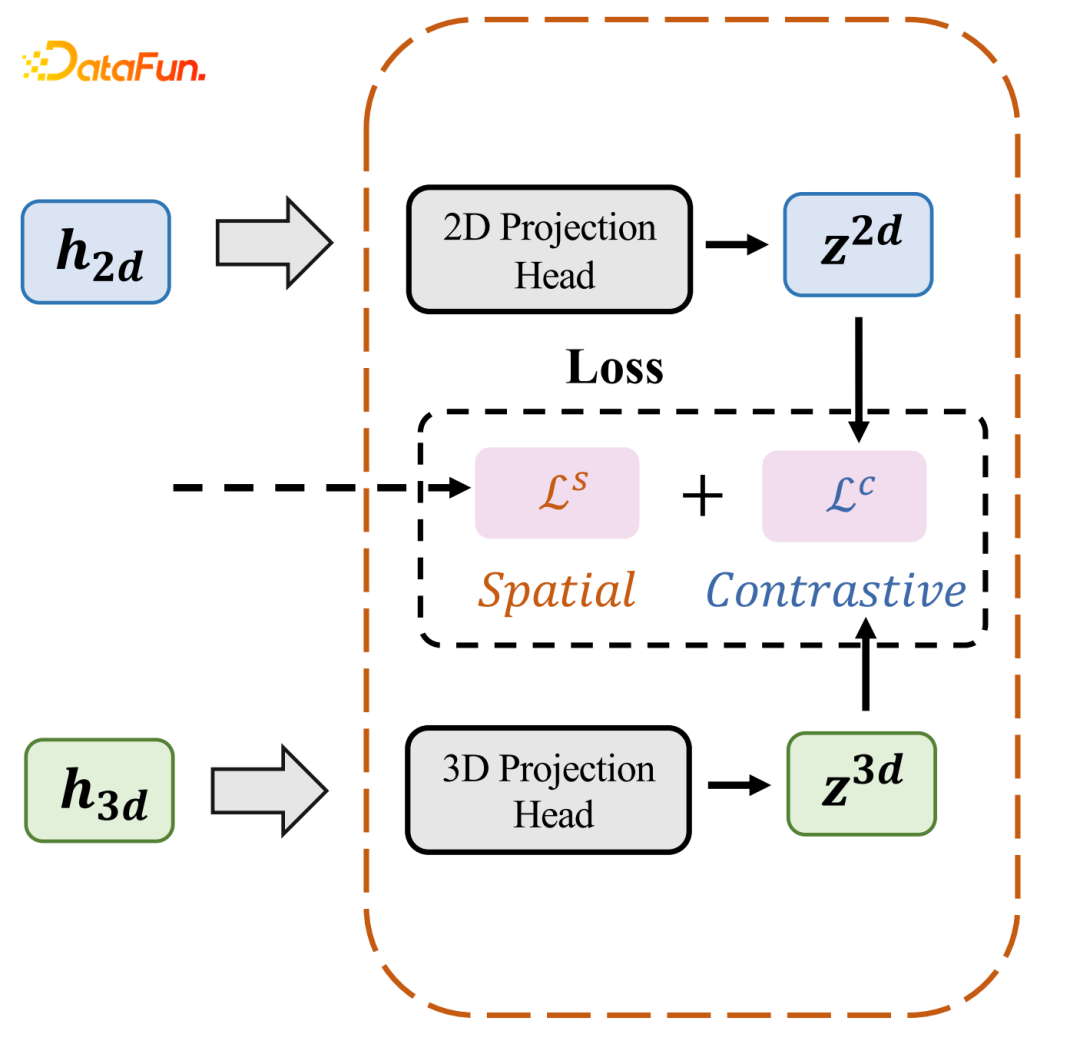
- Overall Framework for GeomGCL
主要分两部分,上一部分是用2D view graph来学习分子表征,另外一个是用3D view graph来学习分子表征,用的方式跟前面介绍的方式是类似的。第二个就是我们引入了contrastive learning的方式,来学习两个节点之间损失函数,来训练和优化模型。
- Geometry-based RBF Encoding
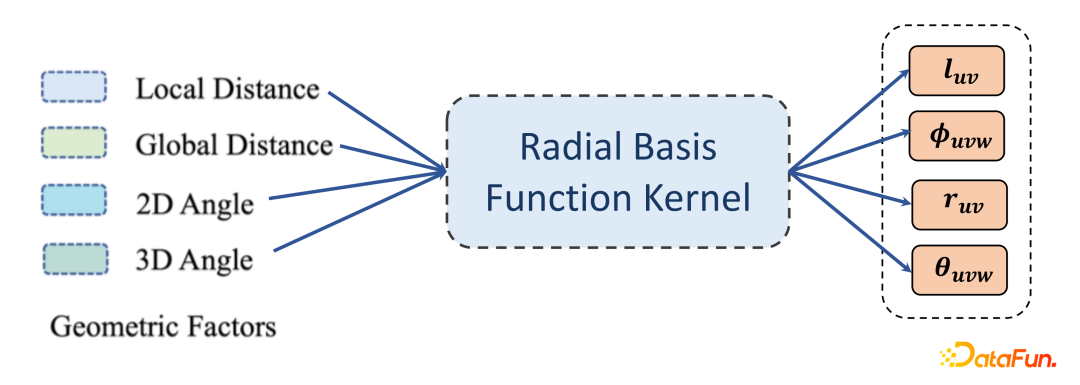
- Adaptive Geometric Message Passing Scheme
-
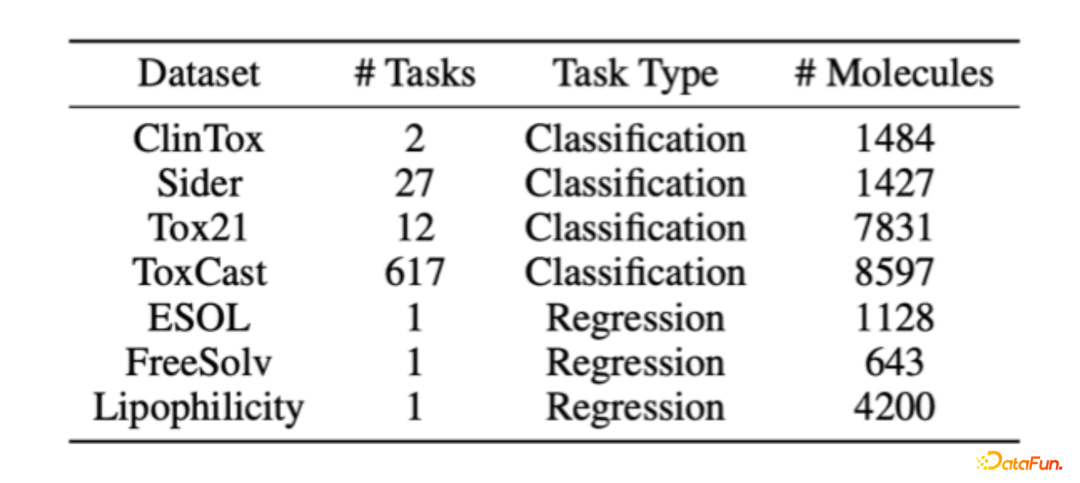
Dataset
-
Baselines

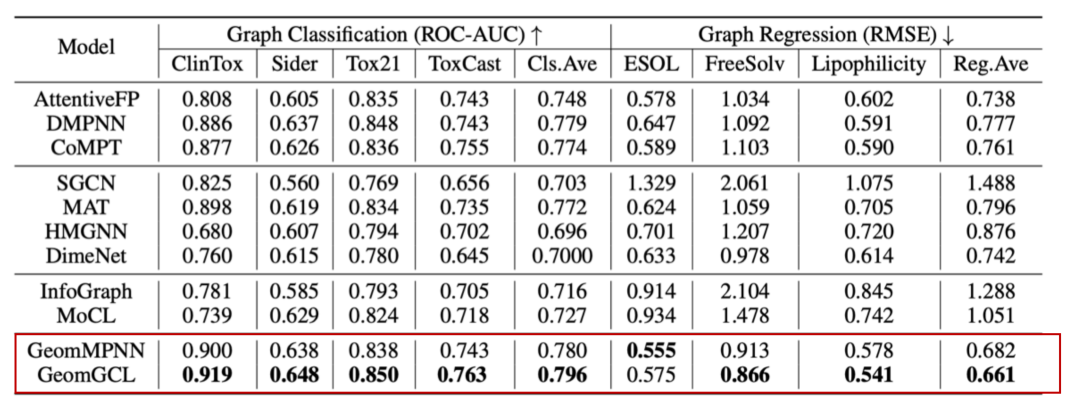
- Comparison with baselines
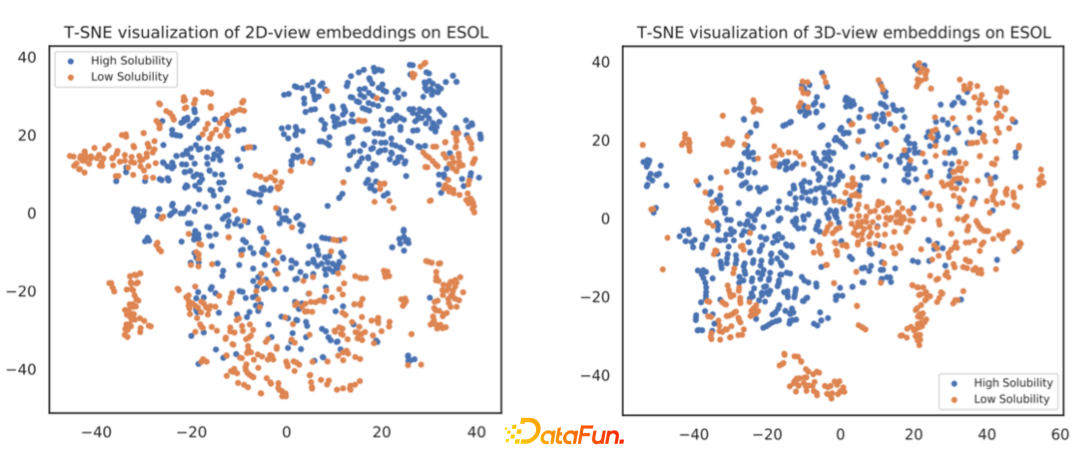
- Summary of our work
这个工作,我们设计了一种dual-channel的Geometric Message Passing的方式来学习同一个分子在2D和3D view下结构上的信息;进而通过contrastive learning的方式来学习小分子的表征。此外,我们通过实验的方式做了不同的downstream task来验证实验结果的有效性。2. 空间结构增强的分子表征学习参考文献:"Geometry Enhanced Molecular Representation Learning for Property Prediction”; Nature Machine Intelligence 2022在此基础上简单讲一下在分子预训练方面的一些工作:
- 化合物表征模型GEM
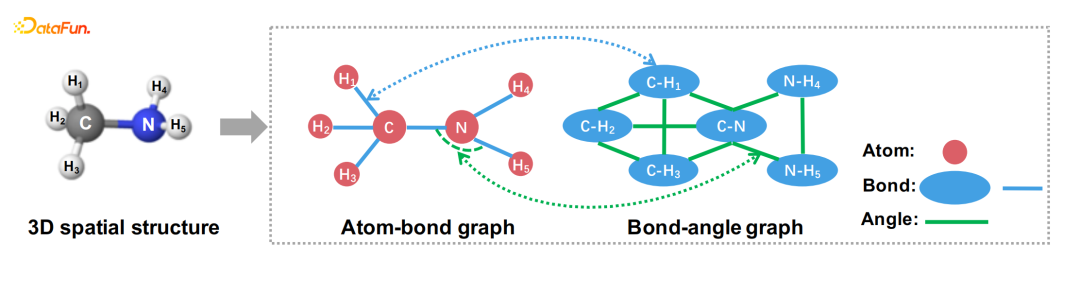

**
**
|分享嘉宾|
**
**

周景博 博士
百度研究院 资深研究员
周景博,现任百度研究院商业智能实验室资深研究员,主要从事数据挖掘和机器学习相关的研究和应用工作, 包括时空数据挖掘、深度几何学习和知识图谱等。2014年从新加坡国立大学获得博士学位,并于2015年加入百度研究院。他目前已经有超过30余篇论文发表在计算机顶级会议和期刊上,包括KDD, SIGMOD, ICDE, AAAI, TKDE和Lancet Public Health,Nature Machine Intelligence等,并常年担任KDD, AAAI, IJCAI, ACL, CIKM, TKDE, VLDBJ等顶级学术会议和期刊的程序委员会委员和审稿人。他作为组委会核心负责人之一承办了KDDCup 2022机器学习竞赛并担任出题人。