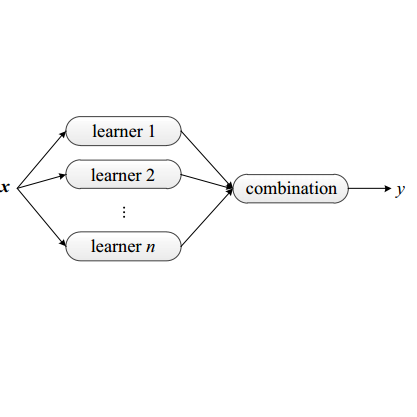
Machine learning force fields (MLFFs) are a promising approach to balance the accuracy of quantum mechanics with the efficiency of classical potentials, yet selecting an optimal model amid increasingly diverse architectures that delivers reliable force predictions and stable simulations remains a core pratical challenge. Here we introduce EL-MLFFs, an ensemble learning framework that uses a stacking methodology to integrate predictions from diverse base MLFFs. Our approach constructs a graph representation where a graph neural network (GNN) acts as a meta-model to refine the initial force predictions. We present two meta-model architectures: a computationally efficient direct fitting model and a physically-principled conservative model that ensures energy conservation. The framework is evaluated on a diverse range of systems, including single molecules (methane), surface chemistry (methanol/Cu(100)), molecular dynamics benchmarks (MD17), and the MatPES materials dataset. Results show that EL-MLFFs improves predictive accuracy across these domains. For molecular systems, it reduces force errors and improves the simulation stability compared to base models. For materials, the method yields lower formation energy errors on the WBM test set. The EL- MLFFs framework offers a systematic approach to address challenges of model selection and the accuracy-stability trade-off in molecular and materials simulations.
翻译:机器学习力场(MLFFs)是一种在量子力学精度与经典势函数效率之间取得平衡的有前景的方法,然而在日益多样化的架构中选择一个能提供可靠力预测和稳定模拟的最优模型,仍然是一个核心的实际挑战。本文介绍了EL-MLFFs,一种采用堆叠方法集成多种基础MLFFs预测的集成学习框架。我们的方法构建了一个图表示,其中图神经网络(GNN)作为元模型来优化初始力预测。我们提出了两种元模型架构:一种计算高效的直接拟合模型,以及一种确保能量守恒的物理原理保守模型。该框架在多种系统上进行了评估,包括单分子(甲烷)、表面化学(甲醇/Cu(100))、分子动力学基准(MD17)以及MatPES材料数据集。结果表明,EL-MLFFs在这些领域中均提高了预测准确性。对于分子系统,与基础模型相比,它降低了力误差并改善了模拟稳定性。对于材料,该方法在WBM测试集上实现了更低的形成能误差。EL-MLFFs框架为应对分子和材料模拟中模型选择及精度-稳定性权衡的挑战提供了一种系统性方法。