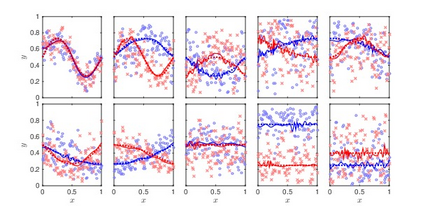
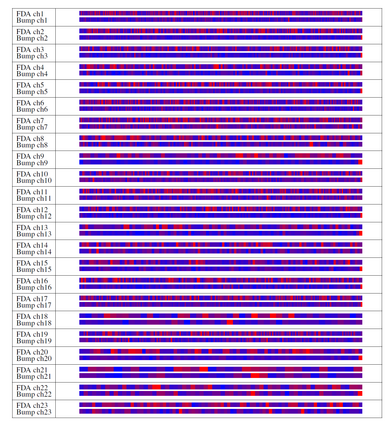
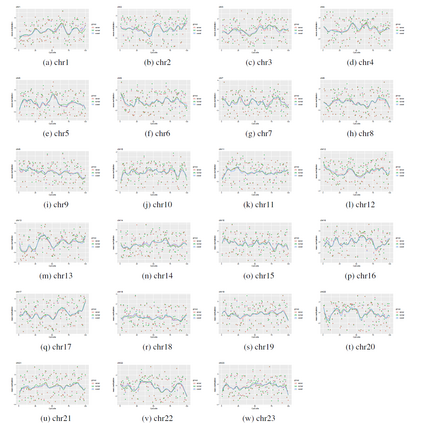
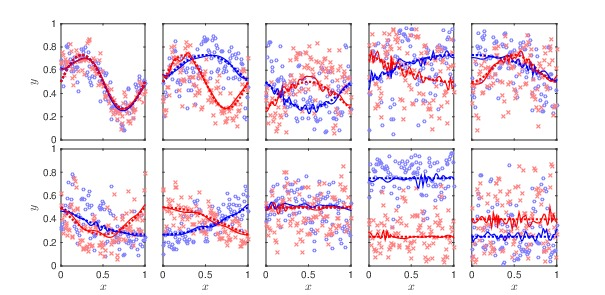
We consider a Bayesian functional data analysis for observations measured as extremely long sequences. Splitting the sequence into a number of small windows with manageable length, the windows may not be independent especially when they are neighboring to each other. We propose to utilize Bayesian smoothing splines to estimate individual functional patterns within each window and to establish transition models for parameters involved in each window to address the dependent structure between windows. The functional difference of groups of individuals at each window can be evaluated by Bayes Factor based on Markov Chain Monte Carlo samples in the analysis. In this paper, we examine the proposed method through simulation studies and apply it to identify differentially methylated genetic regions in TCGA lung adenocarcinoma data.
翻译:我们认为对观测进行贝叶斯功能性数据分析是极其长的序列。将序列分成若干小窗口,其长度可以管理,窗口可能并不独立,特别是当它们彼此相邻时。我们提议利用贝叶斯滑动样条来估计每个窗口的个别功能模式,并为每个窗口的参数建立过渡模型,以解决窗口之间的依赖性结构。每个窗口的人群的功能差异可以由贝叶斯系数根据Markov 链子蒙特卡洛样本进行分析。在本文件中,我们通过模拟研究来审查拟议方法,并应用它来查明TCGA肺癌数据中不同甲基基因区域。