蛋白质磷酸化
是一种广泛的翻译后修饰(PTM),是生物体内一种普通的调节方式,在细胞信号转导的过程中起重要作用。基于数据依赖采集(DDA)和数据非依赖采集(DIA)是基于高分辨质谱的非靶向代谢组学中的常见数据采集模式。
然而,当前的 DIA 磷酸蛋白质组学工作流程面临着一个重大限制,即需要在数据处理之前构建高质量的光谱库。
近日,上海科技大学的科研团队开发了
一个名为 DeepPhospho 的深度学习框架
,
以实现对磷酸肽的 LC-MS/MS 数据的高度准确预测。
通过设计和评估 DeepPhospho 生成的一系列 in silico 文库,证明
DeepPhospho 预测文库优于基准实验 DDA 文库,并实现了更快、更深入的 DIA 磷酸化蛋白质组分析。
该研究以「DeepPhospho accelerates DIA phosphoproteome profiling through in silico library generation」为题,于 11 月 18 日发表在《Nature Communications》杂志上。
DeepPhospho 原理
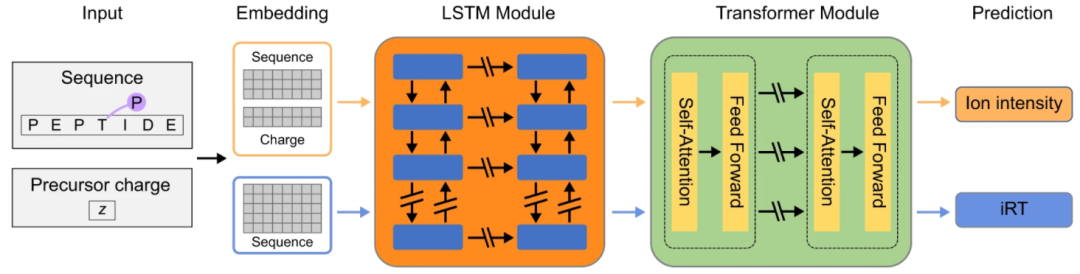
DeepPhospho 的关键组成部分是学习逐渐丰富的肽表示,允许更好地捕获肽的局部和全局结构,以进行细粒度预测。与之前的方法相比,研究人员
采用了一种混合网络设计,该设计集成了两种类型的网络架构来编码肽结构的不同方面。
研究人员开发了一个模块化深度网络,由三个主要子网络组成:用于编码肽的循环网络;用于改进肽表示的 Transformer 网络;用于预测碎片离子强度或索引保留时间 (iRT) 的回归网络。
图示:DeepPhospho 深度学习架构。(来源:论文)
据研究人员表示,
DeepPhospho 是第一个利用 Transformer 预测肽段断裂模式的工作。
为了证明模型设计的优势,进行了消融研究,将模型与 bi-LSTM 或单独的 Transformer 进行比较,并使用两个磷酸化蛋白质组学数据集将 CNN 与 Transformer 相结合。
研究得出:混合模型始终优于那些替代基线,表明 DeepPhospho 能够学习到更好的磷酸肽特征表示,并且 bi-LSTM 和 Transformer 在学习肽表示方面是互补的。
在模型架构测试之后,DeepPhospho 使用四个大规模磷酸化蛋白质组学数据集进行了预训练。然后,研究人员使用 DeepPhospho 对从两个实验室的 Q Exactive HF-X 和 Orbitrap Fusion Lumos 质谱仪获得的其他三个数据集中的磷酸肽进行预测。两个数据集一个通过 DDA,另一个通过 DIA 采集。
经过训练的 DeepPhospho 模型在测试集的实验和预测碎片离子强度之间取得了极好的总体一致性。此外,DeepPhospho 能够在模型训练后对两个数据集进行准确的 iRT 预测。对于第三个数据集,DeepPhospho 对碎片离子强度和 iRT 做出了同样准确的预测。
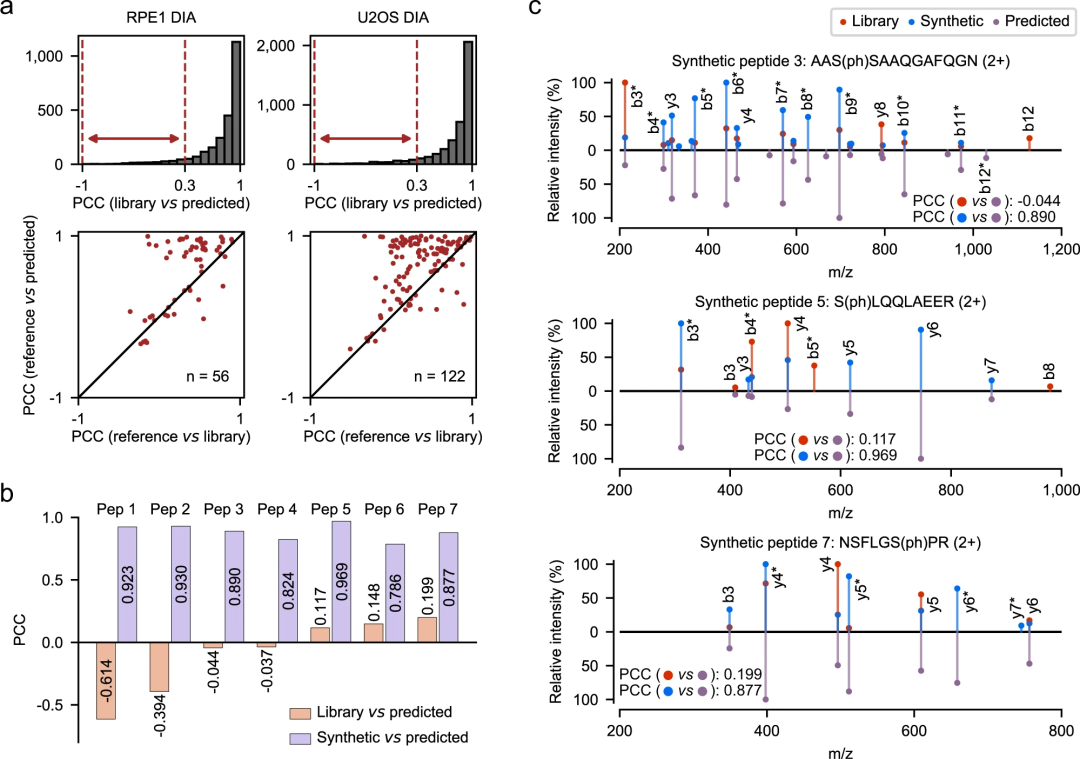
研究人员还将 DeepPhospho 在磷酸肽片段离子强度预测中的性能与最近报道的三个模型进行了比较。在所有情况下,
DeepPhospho 在使用相同的磷酸化蛋白质组数据集进行测试时都优于报告的模型。
图示:DeepPhospho 的 MSMS 光谱预测精确定位了实验库中可能的错误识别。(来源:论文)
总之,
DeepPhospho 能够准确预测磷酸肽的碎片离子强度,在某些情况下,这可以查明实验库中可能存在的错误识别。
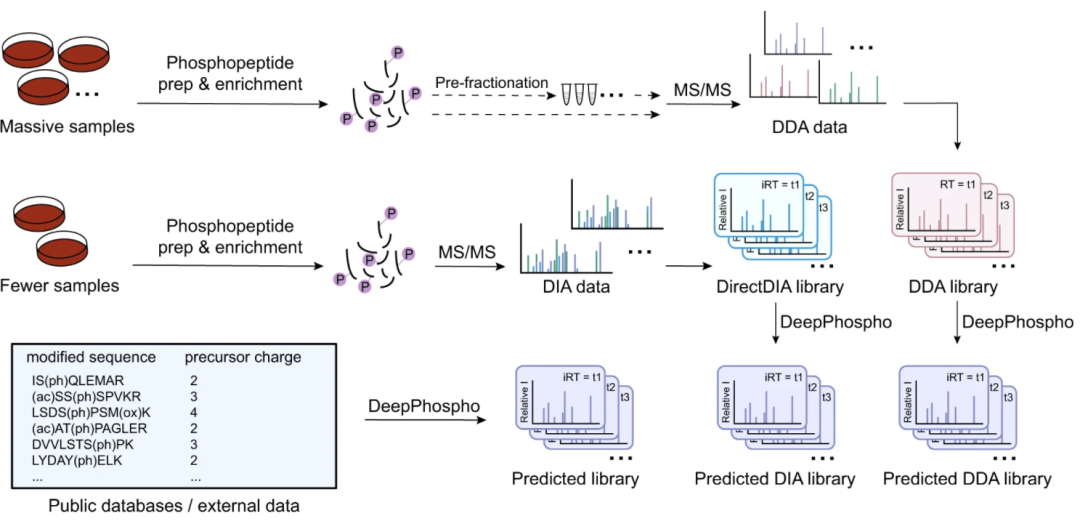
到目前为止,DIA 库的蛋白质组覆盖率仍然落后于广泛的 DDA 库,如果单独使用 DIA 数据,这大大限制了蛋白质组分析的深度。
图示:实验性 DDA 库或直接 DIA 库可以通过 DeepPhospho 转换为预测 DDA 库或预测 DIA 库。还可以从公共磷酸蛋白质组或磷酸位点数据库或外部磷酸蛋白质组学数据生成预测文库。(来源:论文)
为了研究计算机光谱库是否以及在多大程度上可以加深 DIA 磷酸化蛋白质组分析,研究人员设计了六种类型的预测库或混合库,与项目特定的 DDA 库和来自公共磷酸蛋白质组数据库的预测库并行评估。
研究人员使用来自细胞信号研究的 RPE1 DIA 数据集,来评估 DeepPhospho 预测文库在深化磷酸化蛋白质组分析方面的优势是否可以转化为更具生物学意义的场景。
所有五个 DeepPhospho 预测库都通过增加可量化的磷酸肽和磷酸位点的总数而优于广泛的项目特定的 DDA 库(Lib 1)。胜出的是 Lib 6(DIA 和 DDA 的混合体),与 Lib 1 相比,它的磷酸肽和磷酸位点的定量增加了 17.9% 和 14.9%。
图示:使用 DeepPhospho 进行的 DIA 数据分析预测了磷酸化信号研究中的文库。(来源:论文)
为了进一步加深磷酸化蛋白质组的覆盖范围,特别是对于可量化的部分,研究者探索了一种迭代搜索策略。研究发现:对所有重点库的迭代搜索显着增加了可量化磷酸肽的覆盖率,而完全确定的磷酸肽和非磷酸肽的覆盖率几乎没有变化。
总之,证明了
迭代搜索的应用大大促进了可量化磷酸蛋白质组的 DIA 分析,同时不会在数据挖掘中夸大错误发现率或错误定位率。
研究人员表示:「毫无疑问,这样的工作流程将通过增强蛋白质/肽检测,以及减少样本量和仪器时间投资来加速当前的蛋白质组学研究。我们相信 DeepPhospho 和 DIA 磷酸化蛋白质组学工作流程将以各种方式使蛋白质组学和生物学研究受益。」
论文链接:https://www.nature.com/articles/s41467-021-26979-1
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。