DeepMind让AI首次在量子水平描述物质!Nature:化学领域最有价值技术之一
明敏 发自 凹非寺
量子位 报道 | 公众号 QbitAI
现在,AI能在量子层面精准描述物质了!
在最新一期《科学》杂志上,DeepMind构建的神经网络可以预测分子内电子分布,从而计算出分子特性。

这距离DeepMind登上《Nature》封面、解决两大数学难题,仅仅过去了一个星期。
而这一突破对于AI、化学、材料学领域都有重要影响。
一方面,这意味着深度学习在准确模拟量子层面物质上大有前景;另一方面,这对于在纳米尺度探索材料、医学、催化剂等物质都具有重要影响。
DeepMind还表示,他们将开源这一成果给全球科研人员用!
怪不得网友会发出感叹:
DeepMind——YYDS!

《Nature》称这将是化学领域中最有价值的技术之一:
用MLP解决电子相互作用问题
这一次DeepMind解决的问题是密度泛函理论 (DFT)有关。
DFT是一种通过计算分子内电子密度来研究多电子体系电子结构的方法,它可以在量子水平上描述物质,
通过近似的方法,DFT先把复杂的电子相互作用问题简化为无作用问题,再将所有误差另放在一项中,对误差单独分析。
在过去几十年中,它已经成为预测化学、生物学和材料中各种系统特性时最常用的方法之一。
但目前这一方法仍旧存在一定局限性。

一方面,它存在离域化误差。
在DFT计算中,泛函会找到能量最小化时的电子构型来推断分子的电子密度。由此函数误差就会带来电子误差。
大多数已有密度泛函都会错误地将电子密度分布在几个原子或分子上,而不是将其确定在单个分子或原子周围。
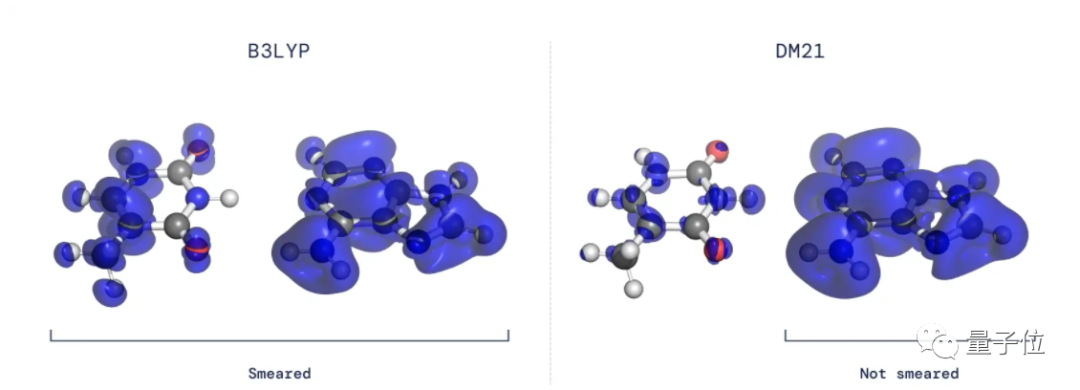
△左图为传统方法,右图为DeepMind提出方法
另一个主要误差来自于自旋对称性破坏。
如果描述结构中的化学键断裂时,现有的泛函会给出一种自旋对称性被破坏的构型。
但是对称性对于研究物理、化学构型有着重要作用,所以当前方法的这一缺陷也就造成了很大的误差。
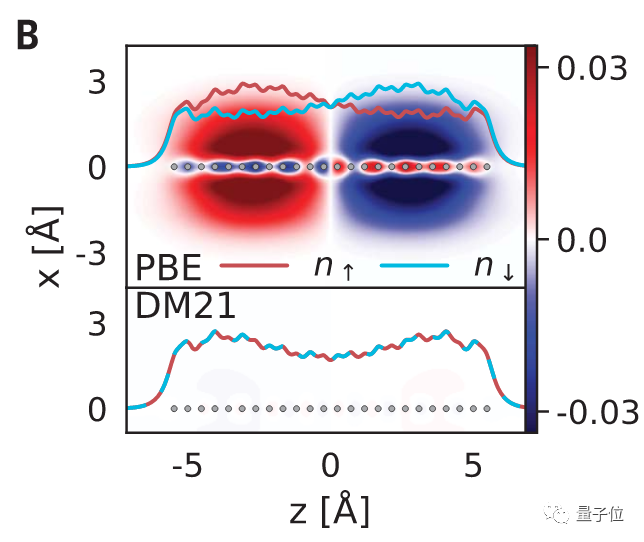
在对比中可以看出,PBE方法打破了自旋对称性。
由此,DeepMind提出了一种神经网络——DeepMind 2021 (简称DM21)。
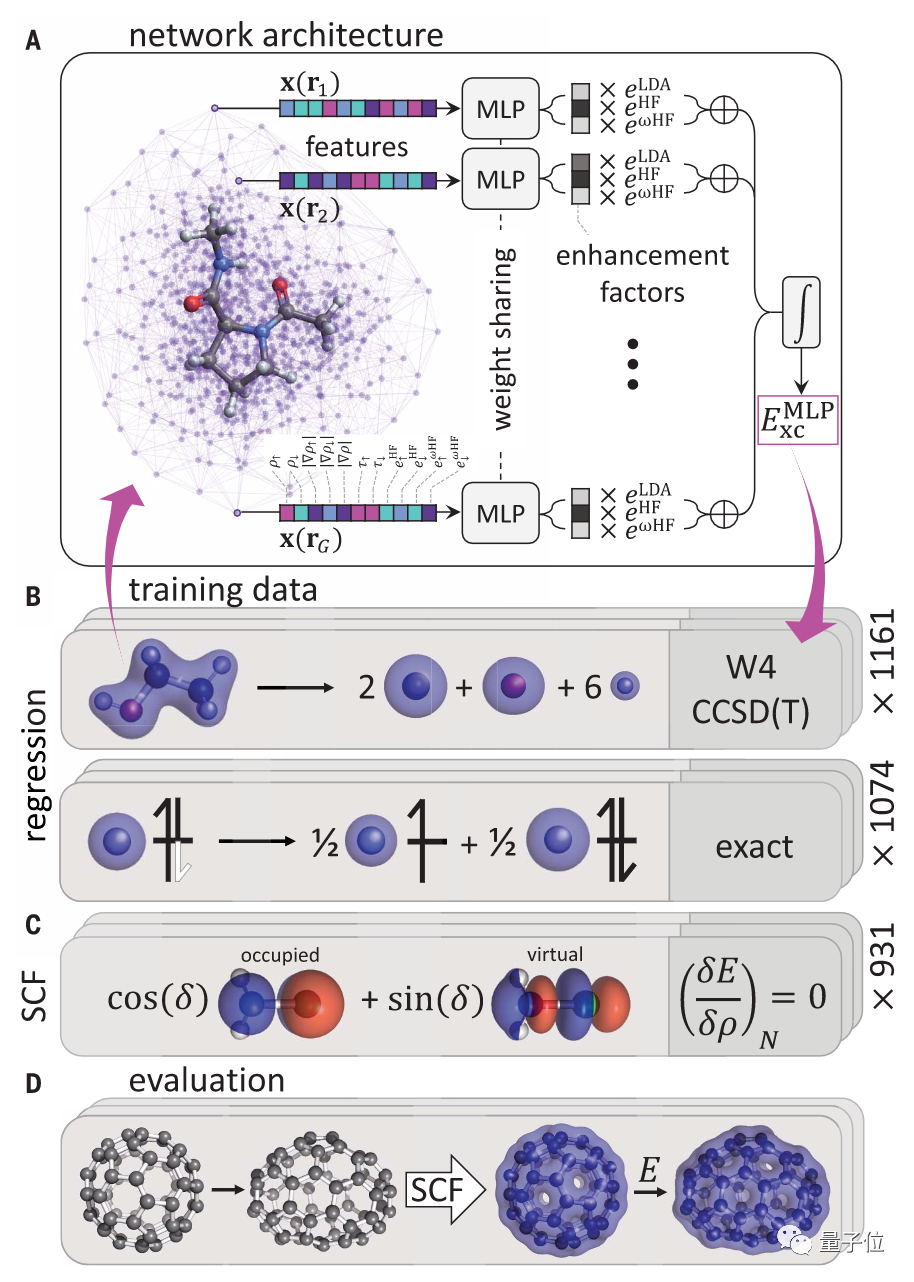
这一框架使用了多层感知器 (MLP),它能映射一组输入向量到一组输出向量。
在向一个权值共享的MLP中输入自旋指数电荷密度等精密化学数据后,它可以预测局部电荷密度的增强值和局部能量密度。
将这些数值整合后,再向函数中增加色散校正DFT。
经过训练后,就可以在自洽计算中部署这一模型。
在具体数据对比中,DM21的误差值都低于传统方法。
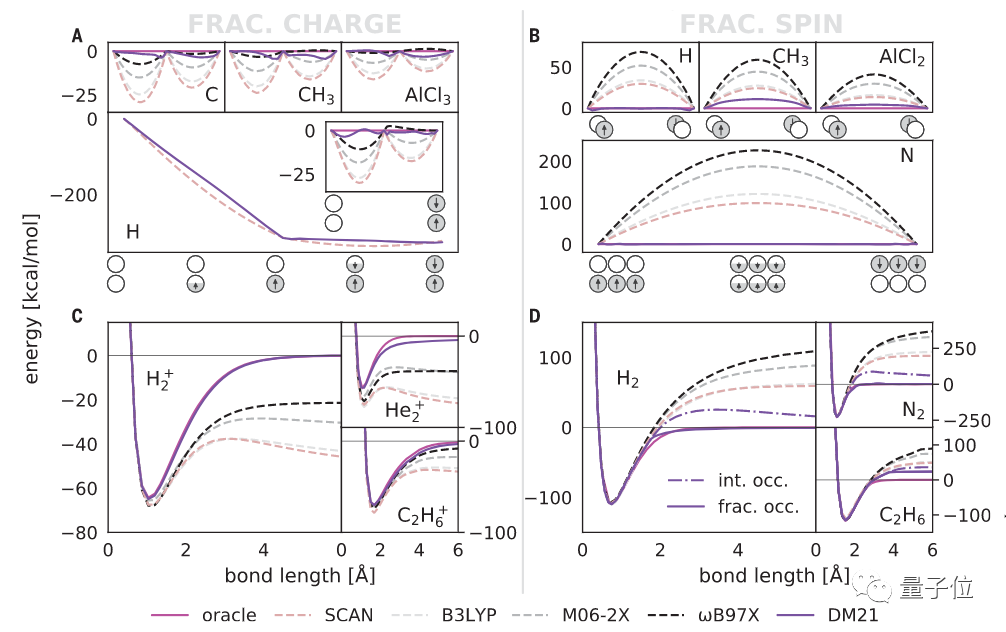
也就是说,DM21可以精准地模拟复杂系统,如氢键链(hydrogen chains)、带电荷DNA碱基对和双自由基体系的过渡态。
实验结果显示,在不同基准(GMTKN55\BBB\QM9)上,DM21的绝对误差值均小于普通方法。
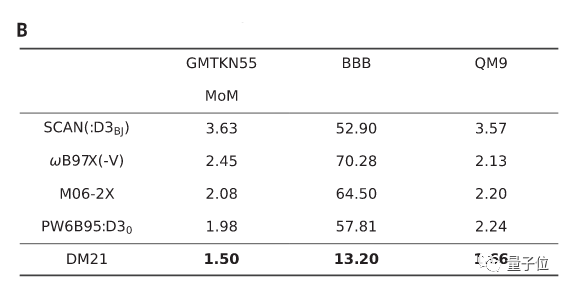
由此不难得出,DM21可以构建出比DFT方法更为精确地描述电子相互作用,深度学习在量子层面精准模拟物质也将大有前景。
已用AI震惊生物界、数学界
本次研究成果的一作为谷歌DeepMind研究学者James Kirkpatrick。
他表示,了解微观现象对于清洁电力、微塑料污染等方面研究都有重要意义。
这对研究人员在纳米水平上探索新材料、药物开发和催化剂等问题,也都有深刻影响。
而这已经不是DeepMind第一次用AI震惊科学界。
在今年,他们用AlphaFold2预测了人类98.5%的蛋白质,一时间震惊生物学界。
不久前,他们用AI突破两大数学难题还登上《Nature》封面,对纽结理论、表示论都产生深刻影响。
论文地址:
https://www.science.org/doi/10.1126/science.abj6511
参考链接:
[1]https://deepmind.com/blog/article/Simulating-matter-on-the-quantum-scale-with-AI
[2]https://twitter.com/DeepMind/status/1469275897614196739
— 完 —
本文系网易新闻•网易号特色内容激励计划签约账号【量子位】原创内容,未经账号授权,禁止随意转载。
2021人工智能年度评选结果揭晓
伴随着产业数字化、智能化的浪潮,AI技术越来越像生活中的水电煤,以润物细无声的形态深入到大众生活的每一个角落。而背后重要的一股推助力,就来自于越来越成熟的平台化解决方案。
「2021年度人工智能最佳解决方案TOP10」榜单中,各个垂直领域的科技头部企业们,正在基于自身的平台实力,支持技术下沉,推助传统行业数智化升级,打造出了一条前沿技术能够被最广泛应用的通途:

p.s.点击图片/链接查看完整榜单:2021人工智能年度评选结果揭晓!AI落地最佳参考在此奉上
点这里👇关注我,记得标星哦~
一键三连「分享」、「点赞」和「在看」
科技前沿进展日日相见~




