【优博微展2017】田昆:基因序列和蛋白质序列分类问题研究


田昆:2017年清华大学优秀博士学位论文二等奖获得者
基因序列和蛋白质序列分类问题研究
Classification of Gene and Protein Sequences
作 者:田昆
指导教师:丘成栋
培养院系:数学科学系
学 科:统计学
读博感言:珍惜时光。
目前生物信息学的研究主要针对基因和蛋白质数据,包括序列、结构等方 面的分析研究。然而,由于数据巨大的规模和极高的复杂性,如果没有有效的 算法,就会导致求解过程花费大量时间,甚至成为无法解决的难题。通过序列 构建的进化树,能进行多个生物物种的序列的同源性分析,从而判断物种间的 发育关系和进化程度。传统的系统发生树构建方法是基于多序列比对,然而这种方法计算时间长,对于高分子大序列很难用它进行比较分析。
我们从不同角度引入序列分析的两种方法――自然向量法和Yau-Hausdorff方法。其中自然向量法提取序列的统计特征,用高维向量表示序 列,而 Yau-Hausdorff方法通过几何图形或点集表示序列。通过大量数据测试, 两种方法都能快速有效地完成序列比对的任务,构建系统进化树,获得与生物学一致的结果,验证了我们的方法的极高准确性和稳定性。
文中主要介绍Yau-Hausdorff方法,它在考虑所有可能的平移和旋转的条件下来完成对两条基因或蛋白质序列图形表示的最优匹配。这种方法的复杂度比目前所有其它的二维最小Hausdorff距离算法的复杂度都低。基于以下两个重要工具,Yau-Hausdorff方法可以用于测定基因序列或蛋白质序列的相似性:
Yau-Hausdorff距离和序列的图形表示。这种序列的图形表示方法保存了序列所有的信息,并且Yau-Hausdorff距离可以严格证明它的确是一个度量,所以Yau-Hausdorff方法能够精确地度量序列之间的差异。
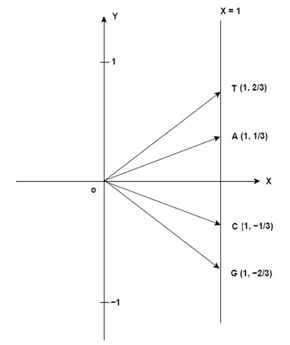
选择四个二维向量来表示四种碱基A,C,G,T
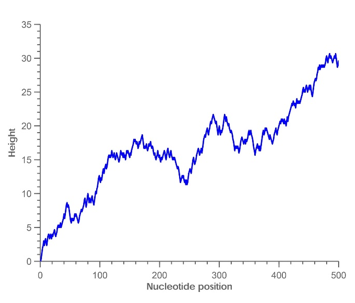
一条长度 500bp的人类线粒体基因序列X93334的图形表示
1、提出了Yau-Hausdorff度量,可以有效判定高维折线的相似关系。
2、发展出一种将抽象序列转化为高维折线的新的图形表示方法,与前述度量结合,导出Yau-Hausdorff算法,可高效的应用于序列分析,且适用范围更广。
3、将上述结果成功应用到基因和蛋白质序列分析,获得与生物学一致的成果。
1、Kun Tian, XiaoqianYang, Qin Kong, Changchuan Yin,RongL. He and Stephen S.-T. Yau, Two dimensional Yau-Hausdorff distance with applications on comparison of DNA and protein sequences, PLoSONE, 2015, DOI:10.1371/journal.pone.0136577, 1-19.
2、YongkunLi, Kun Tian, Changchuan Yin,RongL.He and Stephen S.-T. Yau, Virus classification in 60-dimensional protein space, Molecular Phylogenetics& Evolution, Vol. 99(2016), 53-62.
作者:田昆
供图:田昆
编辑:清华大学研究生院 周明坤 严颖巧