题目: Deep Learning for Network Biology
摘要:
生物网络是发现生物系统中从单细胞到群体水平的相互作用和涌现特性的强大资源。网络方法已被多次用于结合和放大来自单个基因的信号,并导致了生物学上的重大发现,包括药物发现、蛋白质功能预测、疾病诊断和精确医学。此外,这些方法在揭示新生物学方面显示了广泛的实用价值,并为湿实验室实验的新发现作出了贡献。这些方法的核心是网络上的机器学习。在网络上进行机器学习的主要挑战是找到一种方法来提取节点之间的交互信息,并将这些信息合并到机器学习模型中。为了从网络中提取这些信息,经典的机器学习方法通常依赖于总结统计(例如,度或聚类系数)或精心设计的特征来测量局部邻域结构(例如,网络图形)。这些经典的方法可能是有限的,因为这些手工设计的特性是不灵活的,它们通常不能推广到来自其他生物体、组织和实验技术的网络,并且可能在实验覆盖率低的数据集上失败。
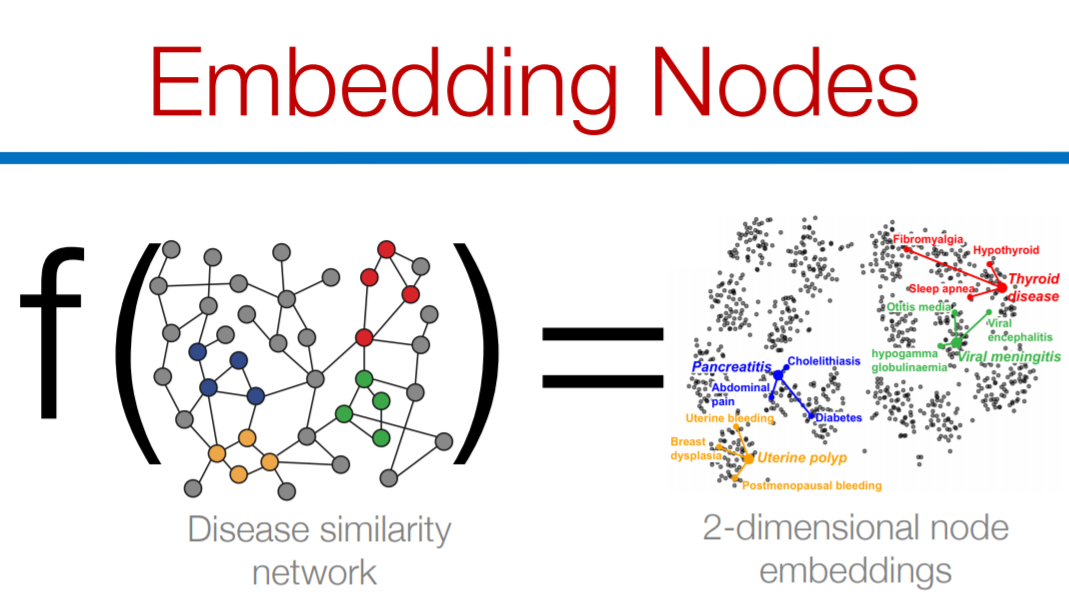
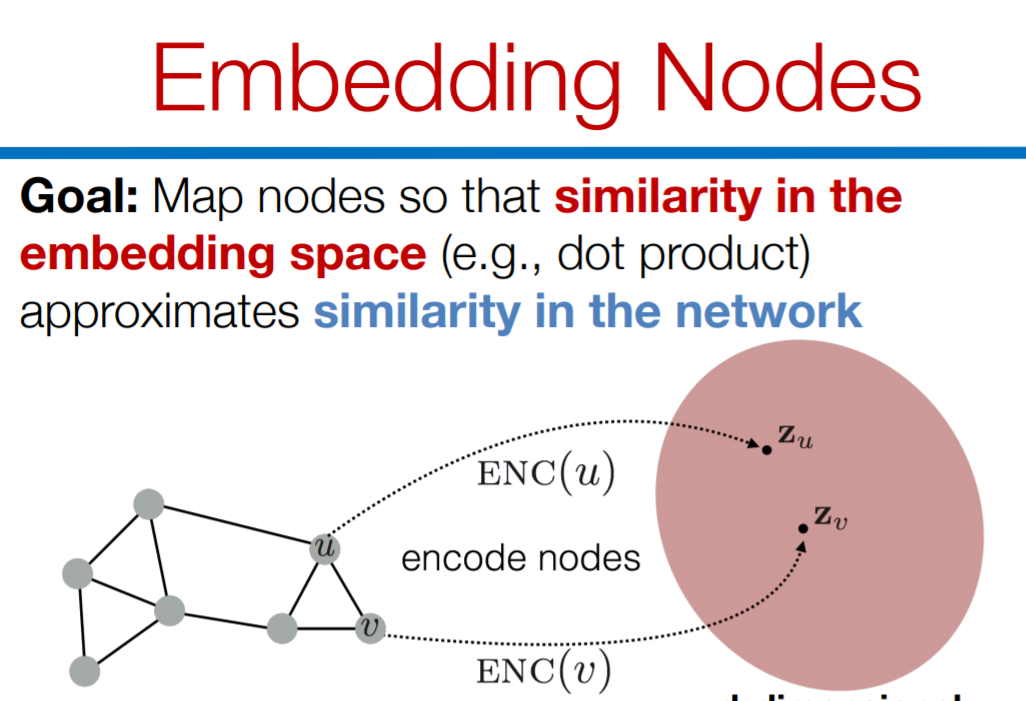
近年来,使用基于深度学习和非线性降维的转换技术,自动学习将网络结构编码为低维表示的方法激增。这些表示学习方法背后的思想是学习一个数据转换函数,该函数将节点映射到低维向量空间中的点,也称为嵌入。表示学习方法已经革新了网络科学的最新技术,本教程的目标是为这些方法打开计算生物学和生物信息学的大门。作者简介:
作者:
Marinka Zitnik是斯坦福大学计算机科学的博士后研究员。她的研究方向是生物医学的网络科学和表征学习方法。2015年,她获得了卢布尔雅那大学的计算机科学博士学位,同时在伦敦帝国理工学院、多伦多大学、贝勒医学院和斯坦福大学进行研究。她在ISMB、CAMDA、RECOMB和BC2会议上获得了杰出的研究奖项,并参与了Chan Zuckerberg Biohub的项目。
Jure Leskovec是斯坦福大学计算机科学副教授,他最近的研究重点是生物和生物医学问题以及网络科学在生物医学和健康领域的应用。2008年,Jure在卡内基梅隆大学获得了机器学习博士学位,并在康奈尔大学学习了一年。他的作品获得了五项最佳论文奖,赢得了ACM KDD杯,并在传感器网络竞赛中获得了冠军。