DeepMind解决50年来生物学重大挑战!破解蛋白质折叠难题!Nature:这可能改变一切

转载新智元
转载新智元
编辑:新智元编辑部
【新智元导读】近日,DeepMind在预测蛋白质结构方面迈出了一大步!公司表示,其已经解决了关键的「蛋白质折叠问题」,这个问题在生物圈已被研究50年之久。马斯克、李飞飞等大佬纷纷点赞!DeepMind这一历史性进展有助于加快药物发现速度,对理解人类生命形成机制至关重要。
刚刚,一个困扰生物学家50年的难题,被AI解决了。
去年年底,谷歌DeepMind推出了一种名为AlphaFold(一个用人工智能加速科学发现的系统,它基于蛋白质的基因序列,就能预测蛋白质的3D结构)的算法。
今天,在有「蛋白质奥林匹克竞赛」称呼的国际蛋白质结构预测竞赛(CASP)上,AlphaFold击败了其余的参会选手,能够精确地基于氨基酸序列,预测蛋白质的3D结构。
其准确性可以与使用冷冻电子显微镜(CryoEM)、核磁共振或 X 射线晶体学等实验技术解析的3D结构相媲美。
DeepMind联合创始人及CEO德米斯·哈萨比斯(Demis Hassabis)表示:「DeepMind背后的终极愿景一直是构建通用人工智能,利用通用人工智能来极大地加速科学发现的步伐,帮助我们更好地了解周围世界。」

谷歌CEO桑达尔·皮查伊(Sundar Pichai)在Twitter上为此次突破点赞。
这是一个跨圈儿的壮举,李飞飞、马斯克等大佬也纷纷点赞:


《自然》杂志更是评论其为「这将改变一切」。
五十年来,蛋白质折叠一直是生物学的巨大挑战。
分子折叠方式变幻无穷,其重要性很难估计。大多数生物过程都围绕蛋白质,而蛋白质的形状决定了其功能。只有当知道蛋白质如何折叠时,他我们才能知晓蛋白质的作用。
例如,胰岛素如何控制血液中的糖水平以及抗体如何对抗冠状病毒,都由蛋白质的结构来决定。
我们都知道,DeepMind以战胜人类而著名,在国际象棋,围棋,星际争霸II和老式的Atari经典游戏中都占据了上风。
但超人游戏从来都不是主要目标,游戏为程序提供了训练场,一旦程序足够强大,就可以解决现实世界中的问题!
蛋白质折叠50年来重大突破,AI破解预测难题
蛋白质折叠50年来重大突破,AI破解预测难题
蛋白质的形状与其功能密切相关,预测蛋白质结构的能力可以帮助我们更好地理解蛋白质的功能和工作原理。世界上许多重大的挑战,比如发展疾病的治疗方法或者找到分解工业废物的酶,从根本上来说都与蛋白质及其所扮演的角色有关。
传统上,得到蛋白质的形状需要花费数年的时间。
从1950年代开始,使用X射线束照射结晶的蛋白质并将衍射光转化为蛋白质原子坐标的技术,确定了蛋白质的第一个完整结构。X射线晶体学已经证明了蛋白质结构的绝大部分。但是,在过去的十年中,低温电子显微镜已成为许多结构生物学实验室的首选工具。
最新的技术进步已使使用低温电子显微镜生成接近原子分辨率的电子密度图成为可能
这些方法依赖于进行大量试验和改进错误,可能需要花费数年的工作时间来完成每个蛋白质结构,并需要使用数百万美元的专门设备来进行试验和验证。
1969年,塞勒斯 · 莱文塔尔(Cyrus Levinthal)指出,用暴力计算法列举一个典型蛋白质的所有可能构型所需的时间比已知宇宙的年龄还要长,他估计一个典型蛋白质有10 ^ 300种可能构型。
1972年,克里斯蒂安 · 安芬森(Christian Anfinsen)在诺贝尔化学奖的获奖感言中,提出了一个著名的假设:

理论上来说,蛋白质的氨基酸序列应该完全决定其结构。
这个假设引起了长达五十年的探索,即仅仅基于蛋白质的 1D 氨基酸序列就能够计算预测蛋白质的 3D 结构。然而,这个假设面临的一个主要的挑战是,理论上蛋白质在进入最终的 3D 结构之前可以折叠的方式数量是一个天文数字。
1980年代和1990年代,尽管早期计算机科学家已经取得了进展,但是从蛋白质的组成中推断结构仍非易事。
为什么预测蛋白质的形状非常困难?
在自然界中,蛋白质是氨基酸链,可以自发折叠成无数令人难以想象的形状,有些甚至在几毫秒之内完成。
为了了解蛋白质如何折叠,DeepMind的研究人员在一个包含约170,000个蛋白质序列及其形状的公共数据库中对其算法进行了训练。在相当于100到200个图形处理单元(按现代标准,计算能力适中)上运行,这种训练需要数周时间。

AlphaFold解决蛋白质折叠问题的方法
AlphaFold解决蛋白质折叠问题的方法
DeepMind 在2018年首次使用最初版本的 AlphaFold 参加 CASP13,在参赛者中获得了最高的准确度,随后又在《自然》杂志上发表了一篇关于 CASP13 方法及相关代码的论文,这篇论文继续启发了其他工作和社区开发的开源实现。
现在,DeepMind开发的新的深度学习架构已经推动了 CASP14方法的变化,使之能够达到前所未有的精确度。这些方法的灵感来自生物学、物理学和机器学习领域,当然还有过去半个世纪许多蛋白质折叠领域的科学家的工作。
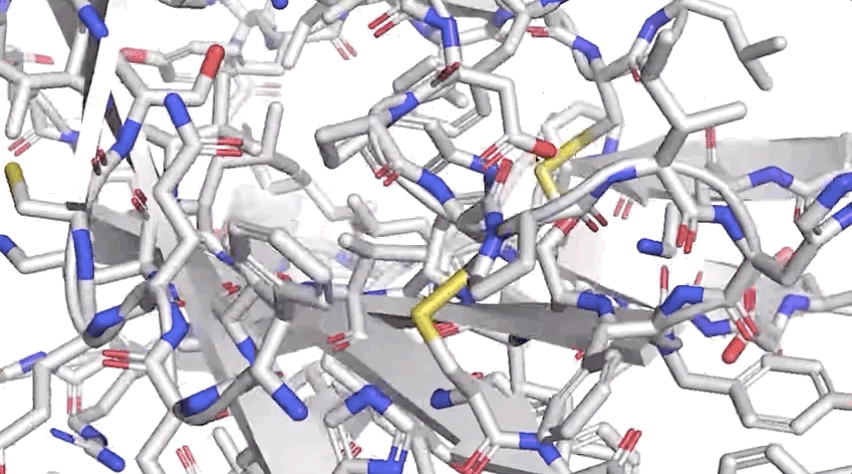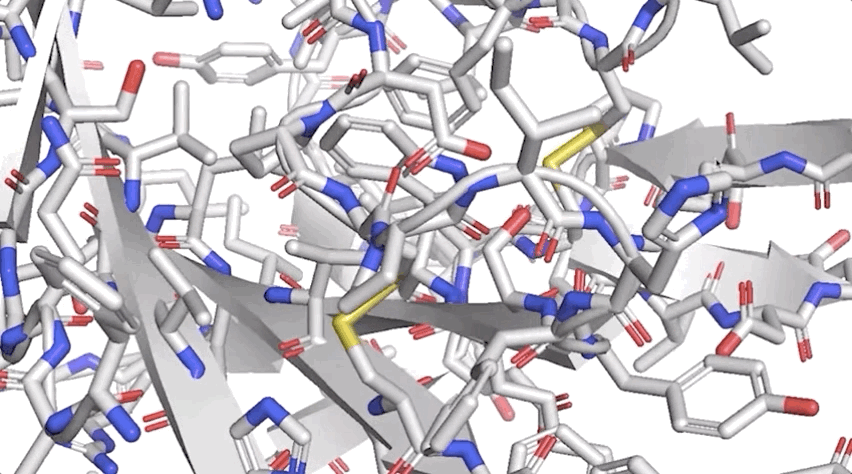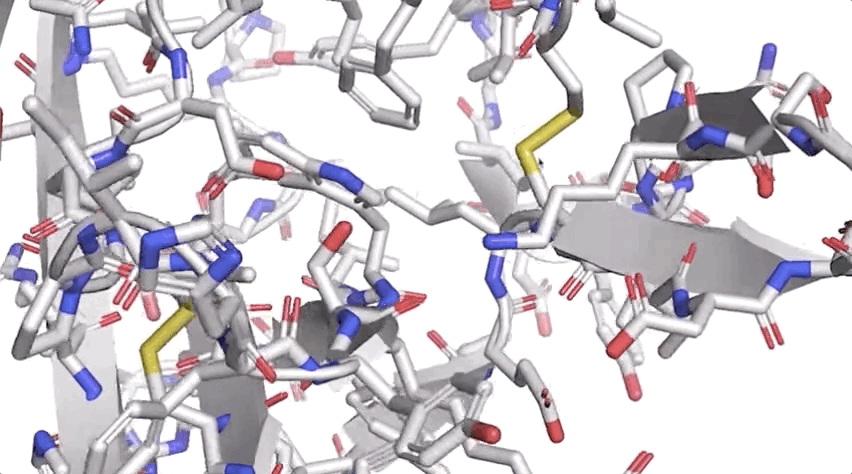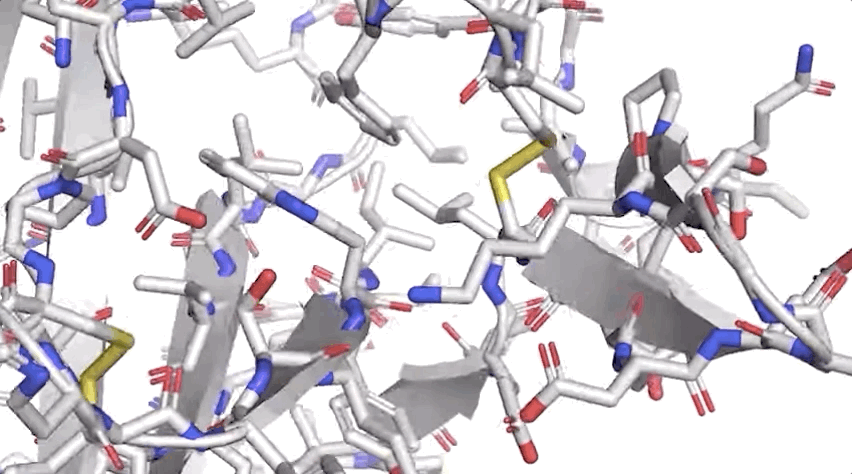
一个折叠的蛋白质可以被认为是一个「空间图形」,其中残基是节点和边连接的。
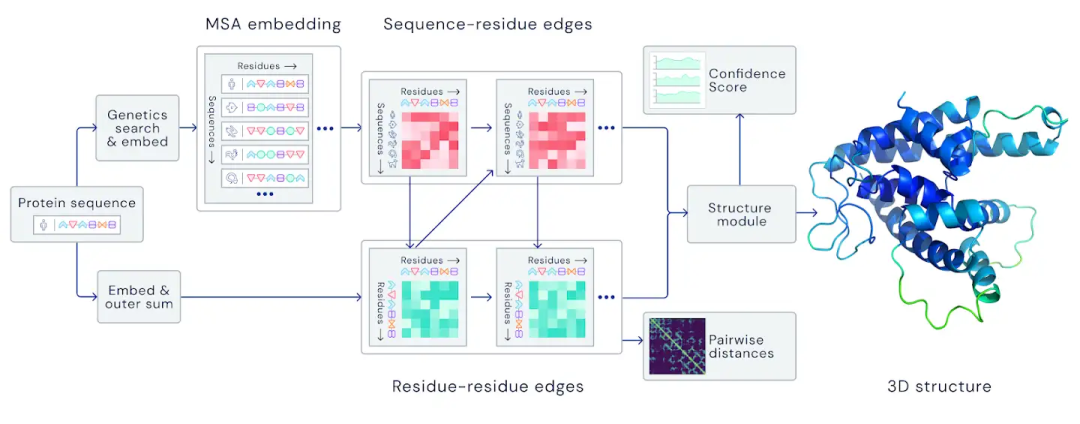
这张图对于理解蛋白质内部的物理相互作用以及它们的进化历史是很重要的。
对于在 CASP14上使用的最新版本的 AlphaFold,研究人员创建了一个基于注意力的神经网络系统,通过端到端的训练来解释这个图的结构,同时推理出它正在构建的隐式图。它通过使用多重序列对齐 (MSA) 和氨基酸残基对的表示来精化这个图结构。
通过迭代这个过程,系统可以对蛋白质的基本物理结构进行准确的预测,并能够在几天的时间内确定高度精确的结构。
此外,AlphaFold 还可以使用内部置信度来预测每个预测的蛋白质结构的哪些部分是可靠的。
这个系统所使用的数据包括来自蛋白质数据库的大约170,000个蛋白质结构,以及包含未知结构的蛋白质序列的大型数据库。它使用了大约128个 TPU v3 (大致相当于100-200个 GPU) ,只训练了几周时间,在当今机器学习领域中使用的大多数SOTA模型中来说是一个相对较少的计算量。
CASP:蛋白质奥林匹克竞赛
1994年,John Moult 教授和 Krzysztof Fidelis 教授创立了 CASP (Critical Assessment of Structure Prediction) 作为两年一次的盲选评估,以促进研究并建立蛋白质结构预测的最新水平。
CASP 是评估预测技术的标准。更重要的是,CASP 选择那些最近才经过实验确定的蛋白质结构(有些在评估时仍在等待确定)作为研究小组测试其结构预测方法的目标; 而这些结构的预测方法并没有提前公布。参与者必须直接预测蛋白质的结构,这些预测随后会在可用时与实验数据进行比较。
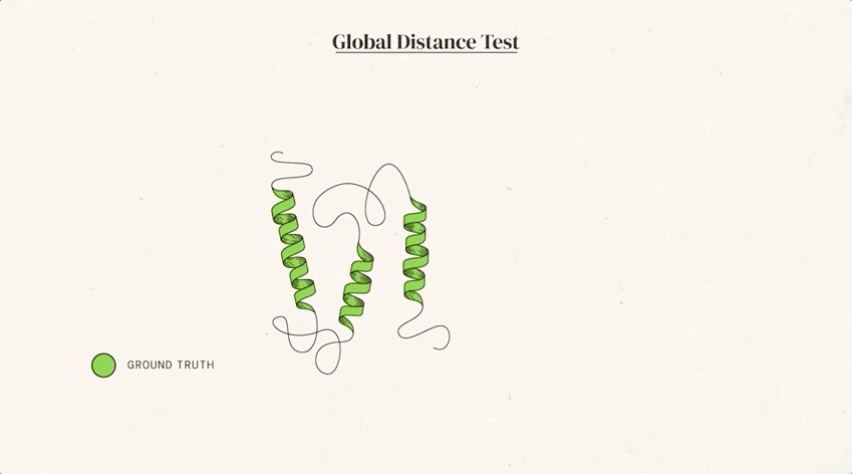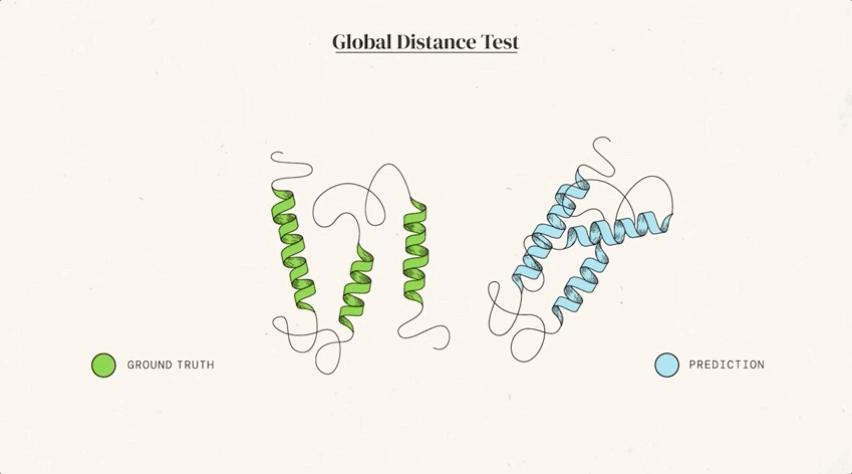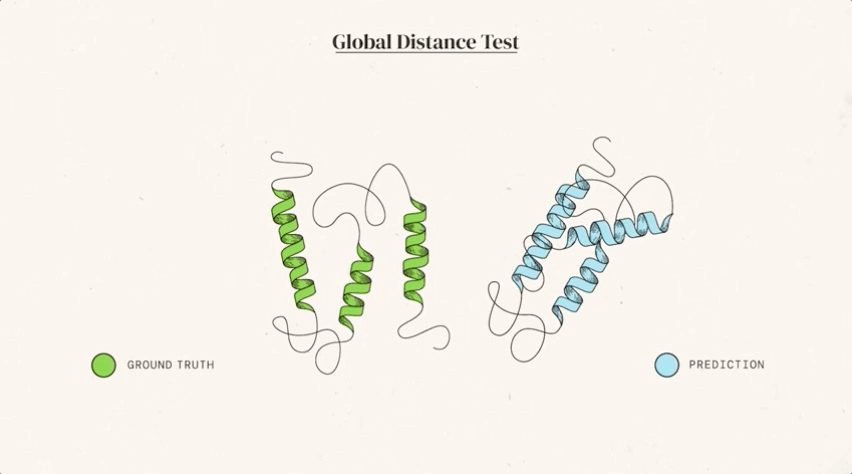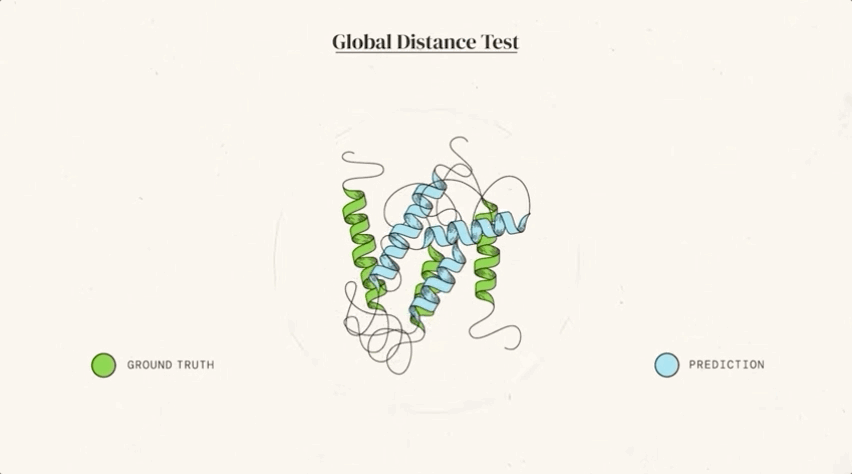
CASP 用来测量预测准确性的主要指标是 GDT(Global Distance Test ) ,其范围是从0-100。简单地说,GDT 可以大致地被认为是氨基酸残基在阈值距离内与正确位置的百分比。根据 Moult 教授的说法,90分左右的 GDT 可以被认为是与实验方法得到的结果相竞争的。

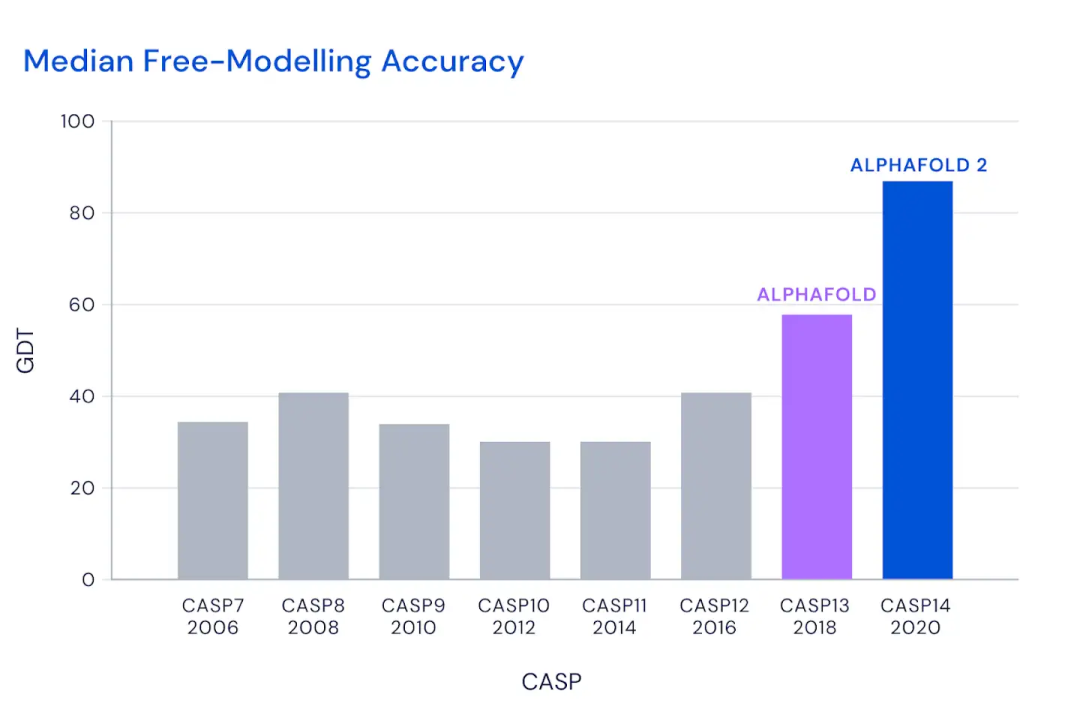
在今天公布的第14届 CASP 评估结果中,最新的 AlphaFold 系统在所有目标中总体得分中位数为92.4 GDT。这意味着预测平均误差(RMSD)约为1.6埃(1埃等于0.1纳米),相当于一个原子的宽度(或0.1纳米),即使对于最难的蛋白质目标,即那些最具挑战性的自由建模分类,AlphaFold 也可以达到87.0 GDT 的中位数。
在接受检验的近100个蛋白靶点中,AlphaFold 对三分之二的蛋白靶点给出的预测结构与实验手段获得的结构相差无几。CASP 创始人 Moult 教授表示,在有些情况下,已经无法区分两者之间的区别是由于AlphaFold的预测出现错误,还是实验手段产生的假象。
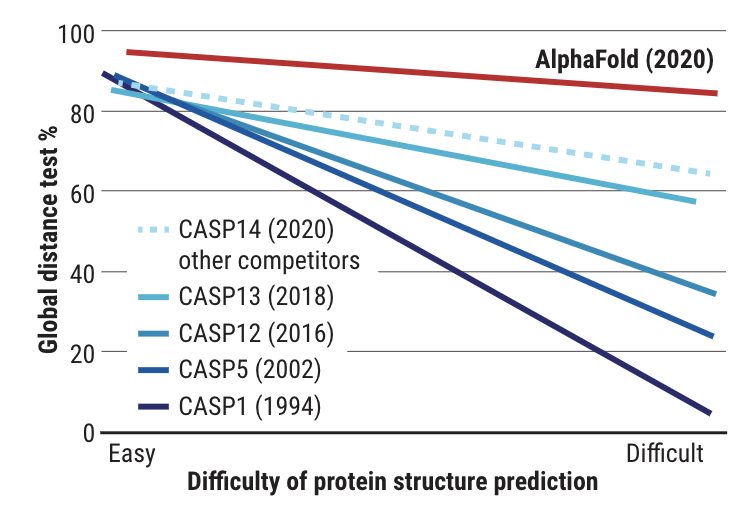
这些令人兴奋的结果为生物学家打开了使用深度学习计算结构预测作为科学研究的核心工具的潜力,DeepMind 的方法可能特别有助于预测重要类别的蛋白质结构,如膜蛋白等。
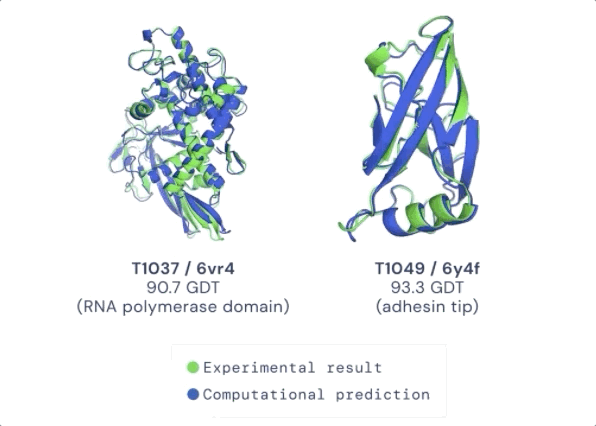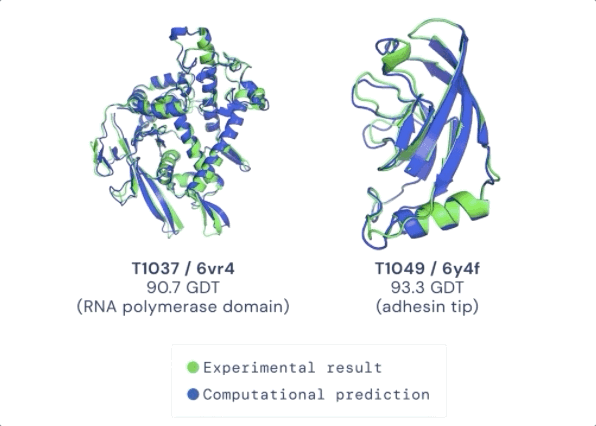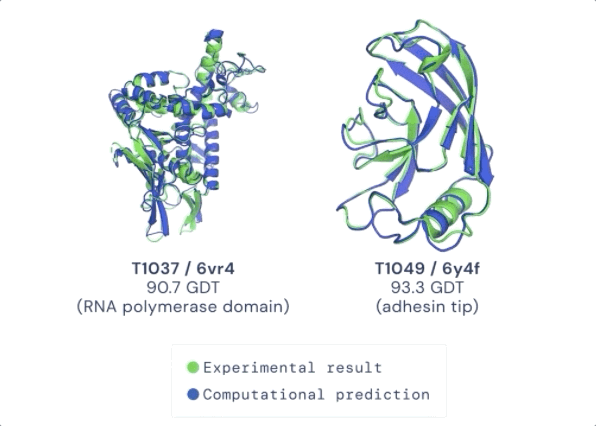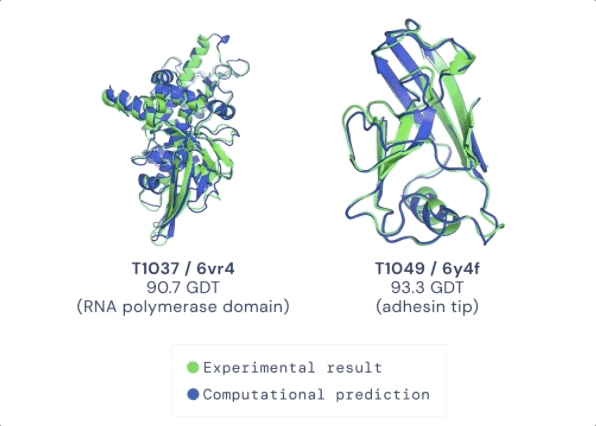
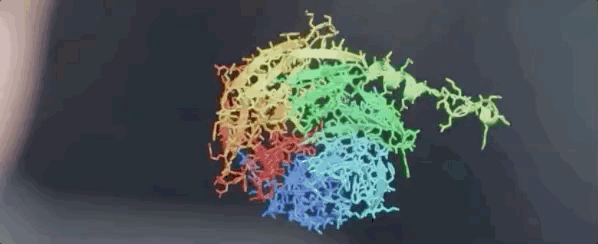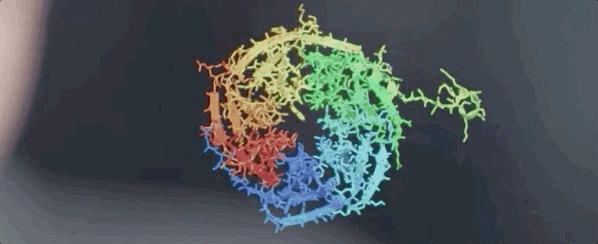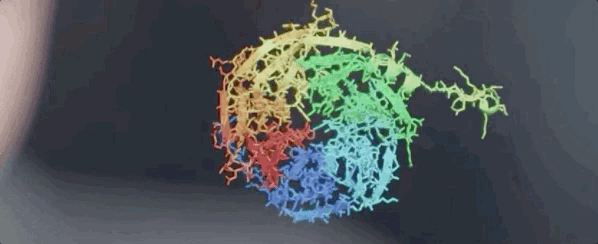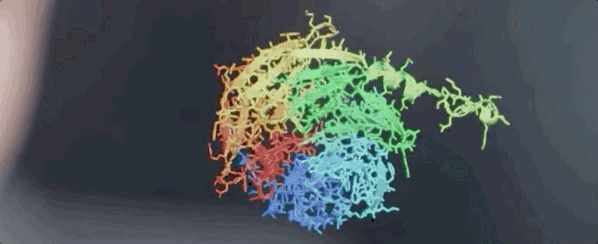
图:ALPHAFOLD 预测了与实验结果相对应的高度精确的结构
历史性突破!AlphaFold将「改变一切」
如果把基因组序列比喻为标识一个人的身份信息,蛋白质的三维结构就是一个人的身形容貌。
预测蛋白质结构变化的重要意义在于,包括癌症、痴呆等几乎所有疾病,都与细胞内蛋白质结构变化相关,如果能够掌握蛋白质结构的变化,将对疾病的预防、治疗等带来重要影响。
通常情况下,识别单个蛋白质的结构需要耗费科学家数年时间。如今,AlphaFold能在几天内提供精确到一个原子的结果。

此举将极大地加速人类对细胞组成部分的理解,对包括新冠肺炎在内所有疾病的研究均有所帮助。
在冠状病毒的结构中,像皇冠一样的刺突称为刺突糖蛋白(Spike Glycoprotein,以下简称 S 蛋白),是结合人体细胞上相应受体的罪魁祸首;E 蛋白是包膜蛋白,将病毒内部的遗传物质包裹起来;还有膜蛋白(M 蛋白)和核衣壳蛋白(N 蛋白)等结构。

三维结构的解析对于新冠病毒致病机理和药物设计具有非常重要的先决意义。
基于AlphaFold的新突破,人类未来也可能更快地发现更先进的新药物。
CASP联合创始人、马里兰大学帕克分校计算生物学家John Moult直言,「这是一件大事,在某种程度上来说,(蛋白质折叠)问题解决了。」
挑战赛评委之一的进化生物学家Andrei Lupas进一步表示,「这将改变医学,这将改变研究,这将改变生物工程,这将改变所有一切。」
知名领域专家Mohammed AlQuraishi发推称;「它们令人震惊——deepmind似乎已经解决了蛋白质结构预测问题。」

谷歌CEO Sundar Pichai在当天也在推特上分享了这一消息并表示:
「DeepMind难以置信的用AI进行蛋白质折叠预测的突破,将帮助我们更好地理解生命的最根本的根基,并帮助研究人员应对新的和更难的难题,包括应对疾病和环境可持续发展。」