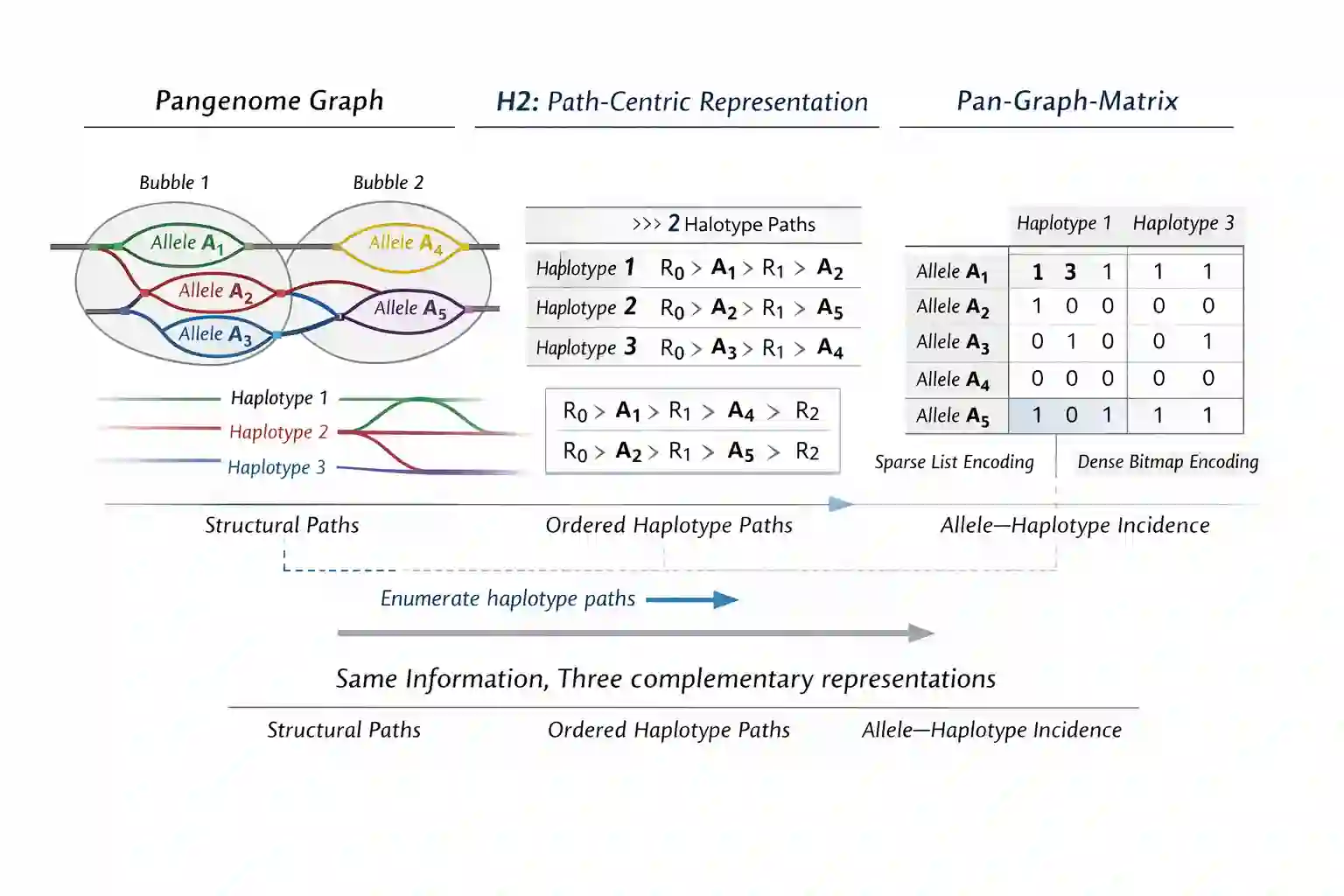
Population-scale pangenome analysis increasingly requires representations that unify single-nucleotide and structural variation while remaining scalable across large cohorts. Existing formats are typically sequence-centric, path-centric, or sample-centric, and often obscure population structure or fail to exploit carrier sparsity. We introduce the H1 pan-graph-matrix, an allele-centric representation that encodes exact haplotype membership using adaptive per-allele compression. By treating alleles as first-class objects and selecting optimal encodings based on carrier distribution, H1 achieves near-optimal storage across both common and rare variants. We further introduce H2, a path-centric dual representation derived from the same underlying allele-haplotype incidence information that restores explicit haplotype ordering while remaining exactly equivalent in information content. Using real human genome data, we show that this representation yields substantial compression gains, particularly for structural variants, while remaining equivalent in information content to pangenome graphs. H1 provides a unified, population-aware foundation for scalable pangenome analysis and downstream applications such as rare-variant interpretation and drug discovery.
翻译:群体规模的全基因组分析日益需要能够统一单核苷酸变异与结构变异、同时在大规模队列中保持可扩展性的表示方法。现有格式通常以序列为中心、以路径为中心或以样本为中心,往往掩盖了群体结构或未能利用携带者稀疏性。我们提出H1全基因组图谱矩阵,这是一种以等位基因为中心的表示法,采用基于等位基因的自适应压缩技术对精确的单倍型成员关系进行编码。通过将等位基因作为一等对象,并根据携带者分布选择最优编码方案,H1在常见和罕见变异中均实现了接近最优的存储效率。我们进一步提出H2,这是一种从相同底层等位基因-单倍型关联信息衍生的以路径为中心的双重表示法,它在保持信息内容完全等价的同时恢复了显式的单倍型排序。利用真实人类基因组数据,我们证明该表示法能实现显著的压缩增益(尤其对于结构变异),同时其信息内容与全基因组图谱保持等价。H1为可扩展的全基因组分析及下游应用(如罕见变异解读和药物发现)提供了统一且具有群体感知能力的基础框架。