中科院赵方庆团队提出环形非编码RNA组学大数据挖掘新技术
2018年1月19日,国际学术期刊 Genome Medicine 在线发表了中国科学院北京生命科学研究院计算基因组学实验室赵方庆团队题为“Reconstruction of full-lengthcircular RNAs enables isoform-level quantification”的最新研究成果。该研究提出全新的环形转录本重构与定量的方法 (CIRI-full),通过环形转录本测序中的反向重叠区特征获得全长序列,既有效解决了环形转录本内部结构的重构难题,也为环形转录本中不同剪切产物的定量提供了新思路。目前绝大多数环形RNA的功能尚未明确,并且现有方法无法提供足够充足序列特征信息,该方法可以帮助研究者更有效地筛选出具有潜在功能的环形可变剪接产物,对环形RNA的功能研究与转录本水平上的差异表达分析具有重要的意义。
在以往的研究中,环形RNA的识别方法主要是利用环形RNA特有结构——反向剪接序列特征进行识别。然而,由于二代测序的读长普遍较短,研究者们虽然可以获得大量的反向剪接位点,却无法高通量获得环形RNA的完整内部结构信息,也无法对不同可变剪接产物进行精确定量。为了解决该问题,赵方庆团队首先提出一个新的环形RNA识别特征:反向重叠区(Reverse overlap)。该特征通常出现在读长较长的环形RNA测序中。与以往的所有识别算法不同,该特征不仅可以用于判断转录本双端测序中的一对测序序列是否来自于环形RNA, 也可以用于判断该序列是否可以覆盖整个环形RNA。研究人员利用该方法从不同哺乳动物的脑组织中获得超过80%的环形RNA的全长序列。
环形RNA的全长重建是对其精确定量的基础。对于拥有多种可变剪接产物的环形RNA,CIRI-full采用蒙特卡罗法模拟来自不同剪切产物的读段在全长序列上的分布,通过梯度下降法,筛选出与最优表达量组合。其中,每个外显子上的测序覆盖度及可变剪接事件都可用于判断预测结果与真实情况的差异。研究人员使用模拟和真实数据验证了这种方法的准确性。对于每个BSJ位点产生多个可变剪接产物的表达数据,CIRI-full不仅可以灵敏地重构其全长序列,并且可以精确地预测其相对丰度。该研究为环形RNA组成和功能研究提供了新视角,呈现了环形转录本更加细致的内部结构,并实现了转录本水平的精确定量,为后续筛选有潜在功能的环形RNA分子提供了重要方法学工具。
该工作主要由赵方庆课题组的博士研究生郑毅和助理研究员冀培丰完成,并获得了国家自然科学基金委、科技部重点研发计划及中国科学院的经费支持。
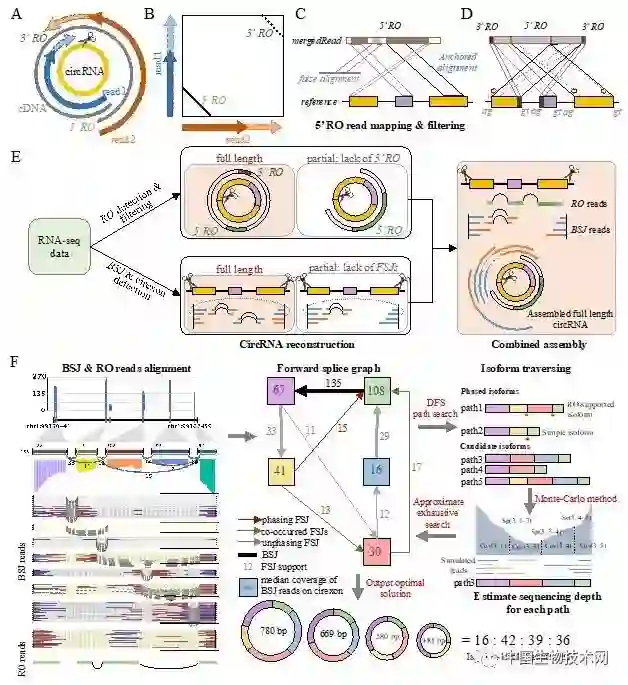
图1. 环形RNA全长转录本的重建和定量方法
中国生物技术网诚邀生物领域科学家在我们的平台上,发表和介绍国内外原创的科研成果。
注:国内为原创研究成果或评论、综述,国际为在线发表一个月内的最新成果或综述,字数500字以上,并请提供至少一张图片。投稿者,请将文章发送至weixin@im.ac.cn。

本公众号由中国科学院微生物研究所信息中心承办
微信公众号:中国生物技术网 回复关键词“热点”可阅读热点专题文章,包括“施一公”、“肠道菌群”、“肿瘤”、“免疫”和“健康”



