npj: Mott物理与过渡金属二元化合物—晶体结构间的牵手
海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

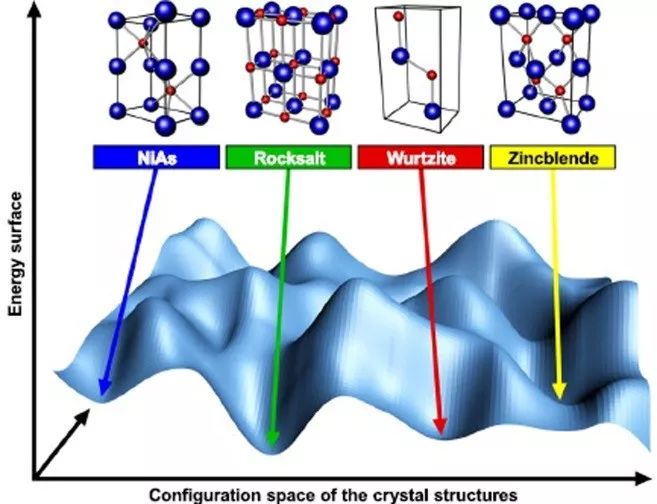
预测结晶材料的基态结构,人们最初认为这是一个无法解决的问题。随着计算总能量方法的有效数值的实现,该预测成为材料研究的活跃领域。如何选择一种晶体结构(稳定或亚稳态)来“制造”固体系统,最终要取决于形成化学键的电子间相互作用。但如何将结构预测方法扩展到强相关材料,之前尚没有报道。
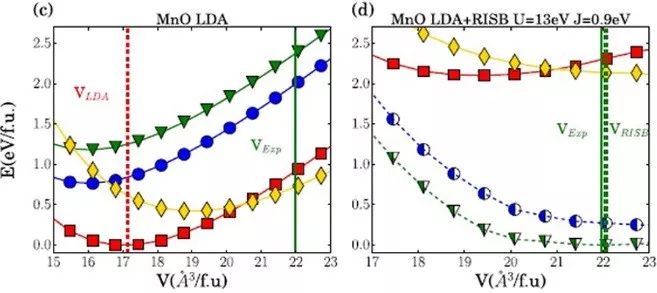
来自丹麦奥胡斯大学的Nicola Lanatà等,将局部密度近似(LDA)与旋转不变的服从-玻色子平均场理论(RISB)相结合,再现了所有化合物的实验已知基态结构,揭示了强相关性影响这些系统中不同晶体结构能量排序的主要物理机制。
他们的主要发现如下:
强电子相关性在所考虑的所有d电子材料中显著影响PES的许多重要特征,如不同多晶型的能量排序和热力学稳定的晶体结构。
描述超出平均场单粒子图像的强电子相关性的有效理论,为我们提供了有效的工具,可用于模拟d电子材料结构的预测研究。
Mott局域化对电荷转移的影响是决定所考虑的所有Mott系统中结构稳定性的关键物理机制,而d-电子共价效应对于预测所有金属系统中的结构稳定性也是必不可少的。
这些结果促进了我们对强相关性调控晶体结构的认识,为将结构预测方法扩展到强相关材料开辟了新的道路,也为预测和研究这些材料体系的亚稳态和多态性铺平了道路。
该文近期发表于npj Computational Materials 5: 30 (2019),英文标题与摘要如下,点击左下角“阅读原文”可以自由获取论文PDF。
Connection between Mott physics and crystal structure in a series of transition metal binary compounds
Nicola Lanatà, Tsung-Han Lee, Yong-Xin Yao, Vladan Stevanović & Vladimir Dobrosavljević
The choice that a solid system “makes” when adopting a crystal structure (stable or metastable) is ultimately governed by the interactions between electrons forming chemical bonds. Here we analyze six prototypical binary transition metal compounds and shed light on the connection between Mott physics and the behavior of the energy as a function of the spatial arrangement of the atoms in these systems. Remarkably, we find that the main qualitative features of this complex behavior in the Mott phase of these systems can be traced back to the fact that the strong d-electron correlations influence substantially the charge transfer mechanism, which, in turn, controls the electrostatic interactions. This result advances our understanding of the influence of strong correlations on the crystal structure, opens a new avenue for extending structure prediction methodologies to strongly correlated materials, and paves the way for predicting and studying metastability and polymorphism in these systems.
扩展阅读
本文系网易新闻·网易号“各有态度”特色内容
媒体转载联系授权请看下方